
A Methodology To Support Particle Investigations In Biopharma Products
By BioPhorum

Particles in parenteral products are a major cause of product recalls, and appropriate management of risk from visible particles is vital to ensure patient safety and product supply. Recent revisions to regulatory standards have prompted the need for a standardized methodology (SM) for investigating the risk of visible particles. This article shares BioPhorum’s recommendations.
Generally, the medical risk to patients from the presence of particles depends on several factors. However, the risk to patients posed by inadvertent injection of sterile, inert particles is commonly minimal, resulting in localized and transient adverse impacts. Consequently, handling findings related to visible particles during and after the manufacturing of parenteral drug product is highly context-dependent.
An occasional occurrence of visible particles in injectables cannot be completely avoided due to current manufacturing process capabilities and the probabilistic nature of particle detection during visual inspections (VI). However, they generally represent an unexpected event that requires an assessment, which may determine that the particle is:
- typical process-related and infrequently occurring
- typical process-related and frequently occurring
- atypical.
In the case of b) or c), further investigations are usually required, often to establish the nature of the particles and root cause and define corrective and preventive actions to support batch disposition, etc. Therefore, adopting a standard approach for more detailed visible particle investigations is essential to achieve a sustained reduction of visible particles. Below is a suggested approach for investigating the occurrence of visible particles and a description of how to evaluate the outcome of the investigation by using a SM.
Investigations related to visible particulates may be triggered by events that occur before, during, and following VI (see Table 1).
Table 1: Common reasons for visible particle investigations.

Generally, the observation of particles during VI leads to an assessment of the following criteria:
- Type/nature of the particle
- Frequency of occurrence for a given product or manufacturing site.
With a), the most common process-related particles originate from equipment or materials used in the manufacturing process. Unfortunately, the current state of technology for the aseptic manufacturing of pharmaceuticals does not allow for a particle-free manufacturing process.
An assessment against b) should consider the number of individual units containing particles (e.g., drug product containers), the number of particles per single unit, and the control limits for the batch. The frequency of occurrence is an important indication of the state of process control.
Investigation Flow
Although several factors can trigger an investigation of the impact of visible particles, investigations generally follow a similar flow (see Figure 1).
Figure 1: Visible particle investigation flow diagram.
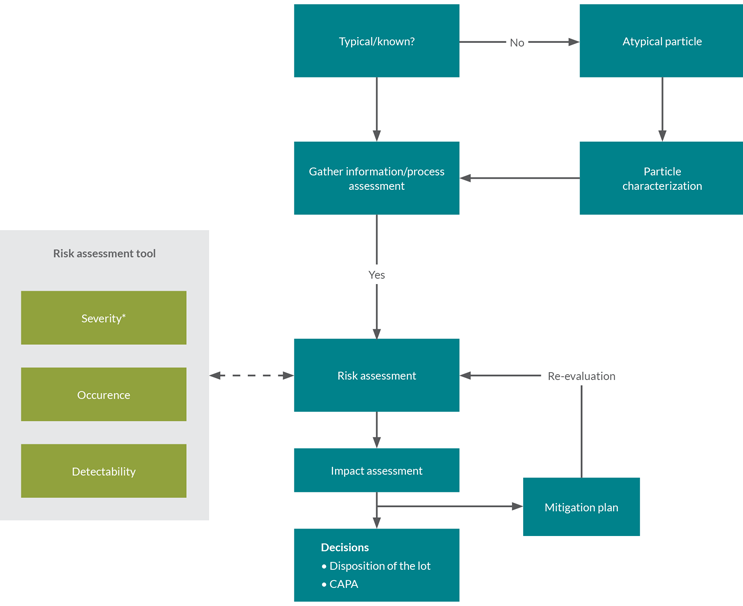
Usually, the first step is to categorize the particle as either typical process-related or atypical. Observation of atypical particles often requires further characterization. The second step is to gather additional data, e.g., the frequency of occurrence of the particle. The next step is to assess the detectability and likelihood of occurrence; severity is also assessed if not already done for the underlying type of particle.
With this information, the investigation can progress to the risk assessment stage.
The risk assessment tool described below may indicate where detectability, occurrence, and severity can be assessed proactively. The tool can be applied prospectively, e.g., for commonly occurring/typical process-related particles. Where a prospective risk assessment is not possible, the risk assessment stage may require further data gathering and a case-by-case application of the tool.
The investigation then proceeds to an impact assessment. Typically, the potential impacts of the findings are considered at this stage, together with a corresponding mitigation strategy and decisions related to batch disposition and corrective and preventive actions.
Risk Assessment Tool
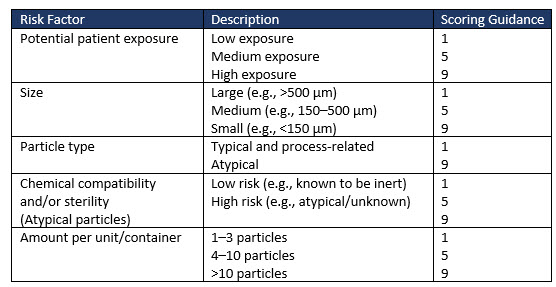
This SM provides users with a decision-making tool to clarify the risks presented by a particle to a batch. It comprises a cross-industry and risk-based standardized approach for assigning a numerical risk value to particles. We propose a simple three-level system for ranking risk, e.g., 1–5–9 or low-medium-high. It is expected that previously identified or typical particles will have already been assessed via the 2018 proof-of-concept guidelines.
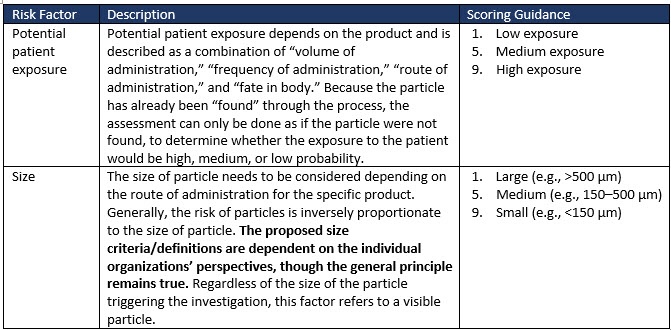
Factors to consider when evaluating the medical risk to patients of visible particles in drug products are exemplified in Table 2 (extract only). Risk factors such as sterility and patient characteristics greatly influence the risk factor ranking adopted by an organization for its drug product.
Risk Factors
Severity Evaluation
Table 2 (extract only) illustrates some of the risk factors and suggested scoring to allow a severity risk to be determined.
Table 2: Examples of risk factors to consider when evaluating the medical risk of particles in drug products (extract only).
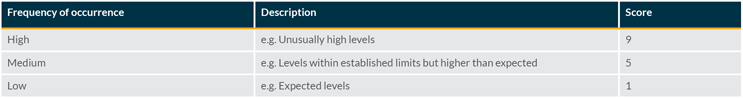
Occurrence Evaluation
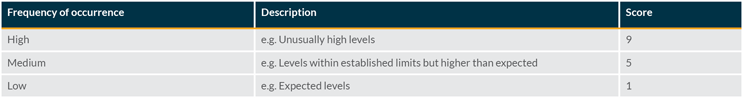
The frequency of occurrence of the visible particle can be evaluated for the specific product being assessed. Factors to consider in this evaluation include actual inspection data from the batch and information gathered from an investigation.
Scoring the occurrence requires a comparison with typical defect rates (average level on standard batches) and an associated investigation limit. This can be done using historical data and may be based on the particle level generated by a standard process.
Once the average level is known and an investigation limit has been established, the following scoring can be used:

Detection Rate Evaluation
The probability of detection is also considered in the tool, which is similar to common failure mode and effects analysis tools. This rating estimates the likelihood that current controls can detect the defect before the product is released to market. As all parenteral products are 100% visually inspected, the detection rating can be estimated from real production data regarding the effectiveness of the inspection process in removing the specific particle being assessed.
The probability of detection of particles is strongly related to the size, type, and color of the particles and may also be influenced by other factors (e.g., the product’s opacity). The following scoring can be used:

The probability of occurrence, severity (impact), and detection are combined to yield the overall risk using the following scoring:
The overall score for severity might be calibrated as:

Risk Analysis And Lot Disposition/Decision
The occurrence and severity are assessed together to calculate the overall patient risk according to the tables below:

Overall patient risk is then assessed against the detectability of the defect according to the tables below:

Relative Risk And Context
The overall risk of visible particles is a risk to a batch or batches and not to a patient. The severity assessed in this tool relates to the risk to the patient (i.e., reacting as if the defect had not been found), but the overall risk assessed relates to an associated batch or batches. This is an important distinction that could introduce confusion for investigators and decision-makers. The existing data suggests that the overall risk to patients of visible particles is low, depending on the product and route of administration.
However, the overall risk of a visible particle to a batch can be similar to the worst-case risk to a batch, i.e., rejection or recall of a batch or batches. This is because a high-risk particle case may indicate an upstream process failure or manufacturing issue. Particle investigations may lead to the detection of another typical particle somewhere downstream of 100% VI due to the probabilistic nature of the visual inspection process. But a particle investigation often leads back to an upstream process failure. Such an event can implicate multiple batches and represents a higher patient risk than a simple particle investigation depending on the upstream process failure mode. This tool helps distinguish these different particle investigations by assessing many factors presenting risk to a batch.
Conclusion
This SM provides a tool to implement a risk-based approach for classifying visible particles to support decision-making during particle investigations throughout clinical and commercial bio/pharmaceutical manufacturing. Through evaluation of the severity of a particle, its rate of occurrence, and probability of detection, it is possible to establish the relative risk of a visible particle to the disposition of a given batch of injectable drug product and provide a framework through which the tool can be used to demonstrate the impact of risk mitigation.
This article is a summary of a recent BioPhorum publication on the topic. To read more, check out the full report, Standardized methodology to support particle investigations and classification in biopharmaceutical parenteral products.