
A Functional History Of Process Validation: Part 2 – The Key To A More Effective Future
By Mark F. Witcher, Ph.D., biopharma operations subject matter expert
Part 1 of this two-part series discussed the history and underlying concepts of process validation (PV), which is required for dealing with the increasingly sophisticated pharmaceutical manufacturing processes necessary for realizing advanced medical therapies.1 As the pharmaceutical industry evolves from chemicals through biopharmaceuticals to cell and gene therapies, the importance of PV is becoming better appreciated as the key to high-quality pharmaceuticals. This article describes how PV concepts can be evolved so the industry can better develop, manufacture, and launch the next generations of biopharmaceutical products.
for dealing with the increasingly sophisticated pharmaceutical manufacturing processes necessary for realizing advanced medical therapies.1 As the pharmaceutical industry evolves from chemicals through biopharmaceuticals to cell and gene therapies, the importance of PV is becoming better appreciated as the key to high-quality pharmaceuticals. This article describes how PV concepts can be evolved so the industry can better develop, manufacture, and launch the next generations of biopharmaceutical products.
As described in part one, PV currently suffers from the following shortcomings that result in a great deal of variation and uncertainty in the approaches used by the biopharmaceutical industry and regulatory agencies to develop, approve, and launch new products:
- Lack of a process definition prerequisite
- Inadequate parameter definitions
- Weak design space description
- Ineffective risk management methods
- Weak control strategy development
- Poor quality by design (QbD) implementation
- Insufficiently designed qualification stage
- Inadequately designed verification stage
- Inability to manage legacy products
The combination of the above limitations has led to substantial confusion and variability in not only the methods companies use to develop products and processes but also in the approaches used to communicate important information to regulatory agencies. The high variability of the process development and validation information with respect to its detail, format, and substance has a significant negative impact on the timing and efficiency of getting products to patients. While complete, precise definitions of the above terms are not required, nor likely desirable, the underlying concepts and their goals should be sufficiently evolved and described to provide effective development approaches and efficient communication within the industry as a whole.
Future Process Validation
If the concepts behind the ICH Q8 terms described in part one are more completely described and understood, PV can be much simpler and more effective. The following briefly summarizes possible modifications to the FDA’s 2011 PV paradigm and supporting ICH Q8 concepts. Additional information and discussion can be found in the cited references.
- Process definition stage – The goals and requirements of the PV design stage can be established in a separate stage that can, for consistency with the current FDA 2011 PV guidance, be designated as Stage 0 – Process Definition. For manufacturing processes, this would include defining the product’s critical quality attributes (CQAs) and especially the process’ performance requirements for consistently making both the CQAs and any possible unknown CQAs.2, 3
- Complete parameter definition – The definition of critical process parameter (CPP) can be expanded to include three different input parameter types: material parameters (MPs), equipment parameters (EPs), and operating parameters (OPs) and one additional output parameter type called process responses (PRs) to clearly differentiate the process’ performance from the process’ CQA outputs. Each of the four parameters has different control strategies that should be clearly defined. All four parameters and CQAs, along with their control strategies, can be defined as part of the design space (DS).2, 3, 4
- Expanded design space – If the process’ inputs and outputs are clearly separated and identified as described above, then a well-structured design space (ws-DS) can be assembled during PV’s design stage using experimental methods under the control of statistical design of experiments (DOE) and risk management methods.5 All inputs and outputs of each unit operation, along with how output quality attributes become the material parameters for the following unit operation, should be clearly identified. The ws-DS can then be used for aligning and communicating the process’ parameters, including their control structures, both internally within process development and with regulatory agencies.2, 3
- Better risk management – Integrating quality risk management (QRM) within the PV stages, particularly the design stage, is critical to the successful development of manufacturing unit operations. PV can be executed by building process flow diagrams, along with a ws-DS, to assemble system risk structures (SRS) for identifying threats to the process that might cause process performance (PRs) and product quality (CQAs) risk consequences. Unacceptable threat-process-consequence risks can be mitigated by adding or changing the manufacturing process unit operations along with their supporting equipment and instruments to reduce the likelihood of the risk consequence occurring. Because the SRS-QRM approach is based on processes, it can be easily used with ws-DS development to build a wide range of effective control strategies. The SRS approach is described in the literature.6, 7, 8, 9
- Well-defined QbD – Rather than defining QbD in impossible-to-use holistic terms, it can be defined as a very specific practical technique as working quality by design (w‑QbD). If w‑QbD is defined as identifying and solving future problems, then it becomes the mechanism that drives the process design function.2,10 One method of building an effective ws-DS for each unit operation is to treat every input parameter as an input threat using SRS-based QRM to guide the process development of each unit operation to control PR and CQA output risks via managing input threats.5, 11
- Better control strategy development – Control strategies (CSs) should be an integral part of the ws-DS. All CSs must meet three goals: control, proof of control, and improve control.12 Every parameter, either specifically or as part of a broader classification, should have a defined CS. Using the input threat, process, output consequence model of the SRS approach, all input parameters can be treated as threats for assessing and appropriately controlling every parameter.2, 3
- Designing the qualification stage – Demonstrating a process works is a very complex requirement best defined during the design stage (Stage 1-2 – Qualification Stage Design) using w‑QbD. Testing the process begins by assembling process performance information during process development and validation that can then be confirmed during process performance qualification (PPQ) lot testing according to a predetermined testing plan. Any equipment or procedural requirements can be included in the Stage 1 effort. The PPQ testing plan should be considerably more comprehensive than the usual product release testing. After the process’ performance has been confirmed, many of the additional tests shown to not yield valuable information can be stopped and normal Stage 3 activities initiated as part of commercial manufacturing.2
- Designing the verification stage – Understanding if a biopharmaceutical process is operating as designed in real time is very challenging. By far the best approach for implementing continued process verification (CPV) is to identify the appropriate OPs and PRs that measure the process’ performance and behavior during the design stage (Stage 1-3 – Continued Process Verification Design) using w-QbD principles.2 When CPV is designed in Stage 1-3, the necessary capabilities and tools for executing CPV can be included in the equipment design, along with real time release testing (RTRT) methods included in the process operating procedures and practices.
- Managing legacy products – The approaches and methods described above can be used when the process for making a legacy product needs to be modified or improved. While the development methods and histories of old products and their processes vary widely, the PV methods described can provide valuable tools for validating existing processes. Using scale-down models, the PV exercise can be completed by expanding the existing process information using the concepts described above. The scope of the legacy product PV project can be defined by either establishing new goals and requirements in an updated Stage 0 or by using a root cause analysis (RCA) to identify the necessary changes and control strategy improvements prior to beginning Stage 1 activities that would also include Stages 1-2 and 1-3 to design the execution stages.

All of the suggested PV enhancements can be summarized in Figure 1. Integrating the above concepts provides the basis for life cycle process development and validation (LPDV). Because development and validation have the same objectives, they can be viewed as simultaneous activities, with all development tasks planned, executed, and appropriately documented to provide a complete validated process created for efficient operation and regulatory approval. This concept is part of ICH Q8’s definition of the design space that concluded with the statement “Design space is proposed by the applicant and is subject to regulatory assessment and approval.” While the path through the LPDV activities may be impacted by financial or timing considerations, the most effective approach combines process development and validation to the extent possible into a single exercise.
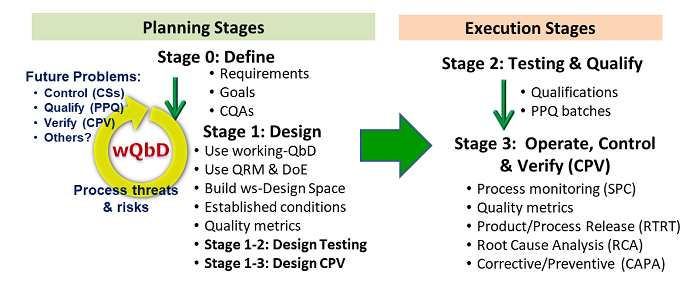
Figure1: The four stages of life cycle process development and validation (LPDV), divided into planning stages and execution stages. The overall paradigm can be generalized to create any process from unit operations to procedures.2
In the case of a legacy process that is experiencing quality problems, the paradigm in Figure 1 can be worked backwards using a root cause analysis (RCA) to identify what CS modifications are required. After the processes are modified to improve control, an abbreviated LPDV can be executed, including Stages 1-2 and 1-3 to qualify, then operate, the improved process. If the process is to be modified, such as to optimize throughput or update equipment, then LPDV can be executed from Stage 0 through Stage 3, building on the existing information from the previous process implementation.
While the discussion above focused on the development and validation of manufacturing unit operations for making products, the LPDV approach can be used for developing or building virtually any process composed of a sequence of operations or activities that take an input and produce an output.
LPDV: A Mechanism For Getting Things Done
Developing and manufacturing biopharmaceuticals requires numerous processes over and above the unit operation (UO) sequence in the manufacturing processes. All processes that make up the three elements of a manufacturing enterprise – manufacturing process (including the UOs), facility, and infrastructure or quality management systems (QMS) – have four basic questions that make up their life cycle:
- What? – Define the process’ goals and requirements.
- How? – Determine how the goals and requirements will be achieved.
- Will it work? – Test the designed process to assure it will work.
- Did it work? – Confirm the process met its goals and requirements.
Answering the four questions is the conceptual foundation for LPDV. The four questions can be used to build virtually any process that is viewed as a sequence of activities (processes) that take an input to produce an output. Using the four questions, either intuitively as a sequence of thought experiments or as a structured, thoroughly documented exercise, provides a mechanism for accomplishing important projects or reaching critical goals. For many simple processes and goals, answering one or more questions may be trivial or obvious, but each one is still important.
The fundamental principles behind LPDV work for any process required for developing and launching pharmaceuticals, including procedures, practices, quality management systems (QMS), equipment, instruments, and even people.13 For all pharmaceutical manufacturing processes, all four questions are extremely important and must be answered and documented completely. The documented answers are the foundational requirements for receiving regulatory approval.
In addition, because LPDV is based on creating and managing processes, it provides a straightforward framework for integrating and using process-based SRS– QRM methods. Thus, LPDV provides an ideal framework for both accomplishing goals and staying out of trouble by using effective risk management methods for controlling risks.
Summary
The key to a bright future for the pharmaceutical industry lies in the use of fundamental principles to tame the overwhelming complexity of new medical products and the process technologies required to make them. LPDV provides a straightforward paradigm for not only building the manufacturing unit operations but also the many processes necessary for developing and manufacturing biopharmaceuticals from procedures, practices, designing equipment, etc. By combining LPDV with SRS-based QRM, all the processes can be systematically developed, operated, and managed using risk analysis methods that identify and control the processes to produce high-quality safe and effective therapeutics.
References:
- Witcher, M. “A Functional History of Process Validation: Part I – A Less than Sufficient Past & Present,” Pharmaceutical Online, https://www.pharmaceuticalonline.com/doc/a-functional-history-of-process-validation-part-a-weak-foundation-0001
- Witcher MF. Integrating development tools into the process validation lifecycle to achieve six sigma pharmaceutical quality. BioProcess J, 2018; 17. https://doi.org/10.12665/J17OA.Witcher.0416
- Witcher, M. F., Why Controlling CQAs Isn’t Good Enough For Gene & Cell Therapies, Cell and Gene Therapy, March 31, 2020, https://www.cellandgene.com/doc/why-controlling-cqas-isn-t-good-enough-for-gene-cell-therapies-0001
- Witcher, M. F., “Phase III Clinical Trials – Ever Wonder Why Some Products Unexpectedly Fail? ISPE iSpeak Blog Pharmaceutical Engineering, Aug. 7, 2019. https://ispe.org/pharmaceutical-engineering/ispeak/phase-iii-clinical-trials-ever-wonder-why-some-products-unexpectedly-fail
- Witcher, M. F., “Quality Risk Management (QRM): Part II – Evaluating the impact of Process Parameters (PP) on Critical Quality Attributes (CQAs) for biopharmaceutical products” BioProcess J, Vol. 15, No. 4, Winter 2016. https://doi.org/10.12665/J154.Witcher.Q
- Witcher, M. “System Risk Structures: A New Framework For Avoiding Disaster By Managing Risks,” Pharmaceutical Online, July 13, 2020 https://www.pharmaceuticalonline.com/doc/system-risk-structures-a-new-framework-for-avoiding-disaster-by-managing-risks-0001
- Witcher, M. “What Managing Personal SARS-CoV-2 Risks can teach us about Managing Pharma Risks,” Pharmaceutical Online, June 12, 2020. https://www.pharmaceuticalonline.com/doc/what-managing-personal-sars-cov-risks-can-teach-us-about-managing-pharma-risks-0001
- Witcher MF. Analyzing and managing biopharmaceutical risks by building a system risk structure (SRS) for modeling the flow of threats through a network of manufacturing processes. BioProcess J, 2017; 16. https://doi.org/10.12665/J16OA.Witcher
- Witcher MF. Estimating the uncertainty of structured pharmaceutical development and manufacturing process execution risks using a prospective causal risk model (PCRM). BioProcess J, 2019; 18. https://doi.org/10.12665/J18OA.Witcher
- Witcher, M. F.; “A Working Definition of Quality by Design (QbD): Understanding its Application to the Development and Manufacturing of Biopharmaceuticals;” BioProcess J; Vol. 13, No. 1; Spring 2014. http://dx.doi.org/10.12665/J131.Witcher
- Witcher, M. F., “Using Quality by Design (QbD) to Build Effective Product and Process Control Strategies Based on a Well-Structured Design Space,” BioProcess J, Vol. 13, No. 2, Summer 2014. http://dx.doi.org/10.12665/J132.Witcher
- Witcher, M. F. “Developing Optimal Pharmaceutical Quality Control Strategies,” Pharmaceutical Online, June 14, 2019. https://www.pharmaceuticalonline.com/doc/developing-optimal-pharmaceutical-quality-control-strategies-0001?vm_tId=2138210
- Witcher, M. F., A Straightforward, Risk-Based Approach to Better Quality Management System Design, Pharmaceuticalonline.com, March 18, 2020 https://www.pharmaceuticalonline.com/doc/a-straightforward-risk-based-approach-to-better-quality-management-system-design-0001
About the Author:
 Mark F. Witcher, Ph.D., has over 35 years of experience in biopharmaceuticals. He currently consults with a few select companies. Previously, he worked for several engineering companies on feasibility and conceptual design studies for advanced biopharmaceutical manufacturing facilities. Witcher was an independent consultant in the biopharmaceutical industry for 15 years on operational issues related to: product and process development, strategic business development, clinical and commercial manufacturing, tech transfer, and facility design. He also taught courses on process validation for ISPE. He was previously the SVP of manufacturing operations for Covance Biotechnology Services, where he was responsible for the design, construction, start-up, and operation of their $50-million contract manufacturing facility. Prior to joining Covance, Witcher was VP of manufacturing at Amgen. You can reach him at witchermf@aol.com or on LinkedIn.
Mark F. Witcher, Ph.D., has over 35 years of experience in biopharmaceuticals. He currently consults with a few select companies. Previously, he worked for several engineering companies on feasibility and conceptual design studies for advanced biopharmaceutical manufacturing facilities. Witcher was an independent consultant in the biopharmaceutical industry for 15 years on operational issues related to: product and process development, strategic business development, clinical and commercial manufacturing, tech transfer, and facility design. He also taught courses on process validation for ISPE. He was previously the SVP of manufacturing operations for Covance Biotechnology Services, where he was responsible for the design, construction, start-up, and operation of their $50-million contract manufacturing facility. Prior to joining Covance, Witcher was VP of manufacturing at Amgen. You can reach him at witchermf@aol.com or on LinkedIn.