
A Case Study In Real-Time Release Testing
By Juan L. Torres, PhD, Biogen
Now is the time to revolutionize how both manufacturers and regulators think about biotech manufacturing in order to proactively respond to the anticipated need for vastly increased productivity and efficiency. In order to continuously improve, manufacturers must work together as an industry and in collaboration with regulators to realize the intent of Quality by Design (QbD) and ICH quality guidelines. Transparency and open communication to make realtime release testing a reality is part of the larger vision of “a maximally efficient, agile, flexible manufacturing sector that reliably produces high-quality drug product without extensive regulatory oversight” (1).
Real-time release testing can be defined as a set of in-process controls that may provide greater assurance of product quality than end-product testing. It leverages real-time analysis, including at-line and on-line measurements of critical quality attributes and process parameters, to ensure product quality. The availability of real-time release testing data at the time of batch manufacturing can also improve operational efficiency and inventory control, reducing the resources needed to test batches following manufacture. Increased product and process knowledge—a fundamental concept of QbD and the ICH quality guidelines—is key to effective real-time release testing.
Typical GMP operations involve performing an extensive set of tests according to approved specifications before the material is released to the market or for further processing. Recent ICH guidelines (ICH Q8, Q9, Q10 and Q11), however, suggest an alternative real-time release strategy to provide assurance of product quality prior to release. Real-time release testing uses the principles of QbD to optimize release and stability testing. A combination of manufacturing process understanding, process control, and product knowledge can be used to demonstrate that the material was made according to GMP.
Despite the potential gains that can be realized from real-time release testing, the industry continues to struggle with implementation and, therefore, has not yet realized the potential benefits. Many questions remain. When and where should tests be conducted on the manufacturing line? Which instrumentation should be used? What associated validation requirements are necessary? Where should real-time release data be recorded? How should on- or in-line analyzers be evaluated during manufacturing? And more critically, what do regulators expect?
Consider the following case study of real-time release testing that leverages historical process understanding and robust quality systems to implement real-time release for a biological drug substance process using a science- and risk-based approach. The models are based on substantial historical data and number of completed manufacturing batches: 64 batches used to develop the model and 40 to validate it.
Real-Time Release in Real World
Using enhanced product and process understanding—an overall awareness of the process based on development studies and real-time data from the manufacturing floor—in combination with quality risk management, the enhanced control approach leverages additional drug substance in-process testing and manufacturing controls in order to eliminate redundant testing. This, combined with full endpoint release testing of the subsequent drug product (which has the same formulation and composition as the drug substance) ensures that the drug substance continues to be manufactured with consistent and controlled quality. Figure 1 shows an example of how real-time release testing and predictive models could be used, not only for overall quality oversight, but for real-time adjustments to optimize process reliability.
Figure 1 Example of Real-Time Release Testing and Advanced Process Control of Drug Substance Manufacturing
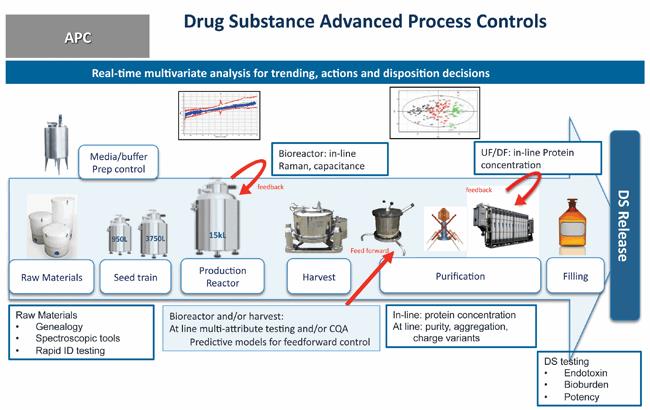
Substance release tests for pH, osmolality, polysorbate 80, protein concentration, purity, charge heterogeneity, aggregation and low molecular weight impurities were performed and controlled at points upstream in the drug substance manufacturing process. The testing points were selected based on the unit operations that either govern or control the generation of the respective product quality attributes. As an additional means to further enhance control of product quality, the action limit ranges for two of the chromatography step yields were narrowed to reflect manufacturing experience obtained over the past five years of production. The tighter action limit ranges further assure the chromatography steps perform as expected and ensure that product quality remains consistent. Safety tests for bioburden and endotoxin, including some rapid test methods, were maintained for the final drug substance release to mitigate potential contamination risks in the drug substance. Based on manufacturing experience, which demonstrated comparability of the analytical results between source drug substance and resulting drug product, testing for appearance, purity (by reducing gel chip) and biological activity was performed only on the drug product, and removed as part of drug substance release testing.
The team overseeing this project submitted it to both US FDA and EU regulators with substantial data and justifications. Meetings were held with the both the US and EU regulators to better understand expectations prior to the submission. These meetings were productive and informative, including open dialogue on expectations. Progress was made through both regulatory bodies and approval has been obtained for EU markets. The team remains optimistic that continued dialogue will open paths to additional markets as both industry and regulators learn more about the possibilities of real-time release testing.
Overall, the development of the data package and submissions for real-time release testing led to open dialogue with both the US and EU regarding expectations and resulted in productive collaborations. This dialogue and interaction between regulators and industry will ultimately define the requirements to move forward into an era where robust process understanding and science- and risk-based approaches are routinely approved. Continued transparency and open communications will pave the way for industry to continuously improve and provide high-quality products.
Note: This article originally appeared in the PDA Letter, a publication produced by the Parenteral Drug Association. March 6, 2017.
Reference
About the Author
Juan Torres, PhD, is Senior Vice- President of Global Quality for Biogen. In this capacity, he has worldwide responsibility over corporate quality, quality control and quality operations at all Biogen production sites.