
21st Century Cures & FDA Implementation: 5 Important Takeaways For Biopharma Companies
By Nancy Bradish Myers, Catalyst Healthcare Consulting, Inc.

Now that the 21st Century Cures bill has been signed into law — following a nearly three- year journey that included a fair number of detours and roadblocks — all eyes in the drug development world are shifting to FDA. To the question “What now?” FDA can answer: A great deal of work.
The almost 1,000-page Cures law is loaded with new requirements and deadlines for FDA to meet. And while the agency scored a “win” on paper in terms of resources, with an additional $500 million over 10 years in FDA innovation projects funds, this new money is not guaranteed. It will be up to appropriators to provide the funds over the 10 years. Thus, FDA must map out its implementation plans without the certainty of new resources. (The agency is adept at implementing massive new laws, but it will be incumbent upon appropriators to provide these funds as intended by Cures sponsors.)
Cures was drafted in parallel with the user fee negotiations. The prescription drug user fee (PDUFA) draft commitment letter contains provisions similar to several of the key priorities for both industry and FDA that have now become law under Cures.
In this article, I will review five major provisions that biopharma, combination product, and advanced therapy companies will need to analyze and incorporate into their business strategies:
- Patient-focused drug development
- Regenerative medicine
- Real-world evidence
- Combination products
- Qualification of drug development tools
Readers will also want to keep in mind that with the new administration coming in in January, the overall implementation plan and speed for Cures could be affected by the priorities of a new FDA commissioner.
Here is my take on what has to happen at FDA now that Cures is law (click image to view a full-sized version of the timeline):
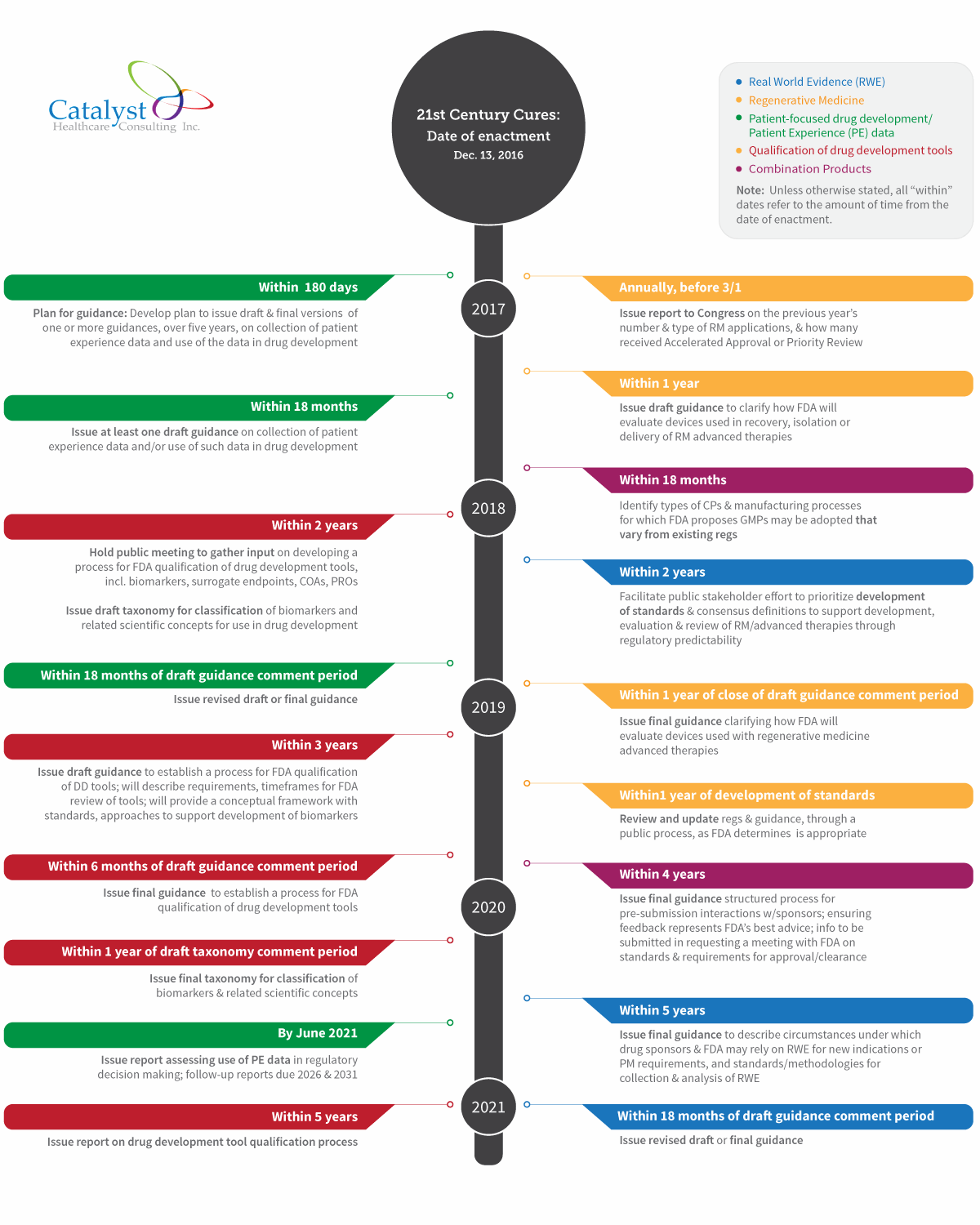
Patient-Focused Drug Development
Of the five major provisions noted, the most immediate deadline falls under patient-focused drug development. Within six months of enactment, FDA must develop a plan for issuing one or more draft and final guidances, over a five-year period.
Cures specifies that these guidances must address, for example, methodological approaches that can be taken to collect patient experience data, methods of facilitating this type of data collection in clinical trials, and methods of developing and submitting a draft guidance related to patient experience data for FDA’s consideration.
The first patient experience draft guidance is due within 18 months of enactment, with the final guidance due within 18 months of the end of the draft guidance comment period. FDA is also required to issue a report within five years, and in five-year increments thereafter (ending in 2031), assessing the use of patient experience data in regulatory decision making.
On an ongoing basis, FDA is also required to issue a statement regarding any patient experience data that was used at the time a drug is approved.
Regenerative Medicine
This section establishes a definition of regenerative medicine (RM) and advanced therapies and allows FDA to consider regenerative therapies as candidates for accelerated approval. FDA is also given a range of new requirements in this area, including:
- Within one year — Issue draft guidance clarifying how FDA will evaluate devices used in the recovery, isolation, or delivery of regenerative medicine advanced therapies, with final guidance due within a year of the close of the comment period
- Within two years — Facilitate a public stakeholder effort to prioritize development of standards and consensus definition of terms to support the development, evaluation, and review of RM advanced therapies, through regulatory predictability
- Within one year of the development of these standards — Review and update regulations and guidance, through a public process, as FDA determines is appropriate
- Annually, prior to March 1 — Send a report to Congress on FDA’s experience in the previous year with regenerative medicine products, e.g., the number and type of RM applications and the number of applications that received accelerated approval or priority review
Real-World Evidence
Real-world evidence (RWE) has been a theme throughout 2016, so its inclusion in the Cures law is fitting.
Within two years of enactment, FDA is required to both:
- Establish a draft framework for a program to evaluate the potential use of RWE to support approval of a new indication, or to satisfy post-market study requirements
- In accordance with this framework, implement the program
The framework, which includes consultation with stakeholders, must describe:
- Sources of RWE, including ongoing safety surveillance, observational studies, registries, claims, and patient-centered outcomes research
- Gaps in data collection activities
- Standards and methodologies for collection and analysis of RWE
- Priority areas, remaining challenges, and “potential pilot opportunities”
At the five-year mark, FDA must issue draft guidance to describe the circumstances under which drug sponsors and FDA may rely on RWE for new indications or post-market study requirements, and standards and methodologies for collection and analysis of RWE for these purposes. This section is similar the RWE provisions in the PDUFA negotiated agreement. RWE revised draft or final guidance is due 18 months after the close of the draft guidance comment period, which means revised/final guidance will be issued slightly beyond the six-and-a-half year point following enactment.
Combination Products
The number of combinations FDA is being asked to review is increasing; for example, FDA received 350 original premarket applications for combination products in FY 2015, a 10 percent increase over FY 2014. Therefore, Cures included several provisions intended to improve coordination among the FDA centers involved in combination product reviews and timeliness of interactions with sponsors. The law includes a section that requires new activities related to GMP requirements and pre-submission meetings with sponsors.
The first deadline requires FDA to identify (within 18 months) types of combination products and manufacturing processes “with respect to which FDA proposes that Good Manufacturing Processes may be adopted that vary from” existing regulations. FDA must take a risk-based approach to identifying such types and variations, and publish a proposed list in the Federal Register. After a comment period of an unspecified length, the agency must publish the final list, and then periodically review the list.
However, FDA has a much longer timeframe — four years — to gather stakeholder input and issue final guidance on combination products. In the guidance, FDA must describe:
- A structured process for managing pre-submission interactions to “provide clarity and certainty” for sponsors
- Best practices for ensuring feedback in these interactions represents FDA’s best advice
- The information required to be submitted in requesting a meeting with FDA to address issues such as standards and requirements for approval or clearance
Other combination products provisions include:
- In reviewing a combination product with an approved constituent part, FDA may require the sponsor to submit “only data or information” that FDA “determines is necessary to meet the standard for clearance or approval,” including any incremental risks and benefits” posed by the combination product, using a “risk-based approach and taking into account any prior finding of safety and effectiveness or substantial equivalence for the approved constituent part relied upon by the applicant.”
- FDA must assign a “primary agency center” to regulate combination products.
- In determining the primary mode of action (PMOA), FDA cannot determine that the PMOA is that of a drug or biologic “solely because the combination product has any chemical action within or on the human body.”
Qualification Of Drug Development Tools
Both FDA and sponsors have been working in recent years to improve the drug development tool qualification process. This issue was also raised, and is addressed by, the PDUFA negotiated agreement.
Under Cures, within two years of enactment, FDA is required to:
- Hold a public meeting to gather input on developing a process for FDA qualification of drug development tools — including biomarkers, surrogate endpoints, and clinical outcome assessments (COAs), including patient-reported outcomes (PROs)
- Issue a draft taxonomy for classification of biomarkers and related scientific concepts for use in drug development, followed by the final taxonomy within a year of the close of the draft comment period
Draft guidance to establish a process for FDA qualification of drug development tools must be issued within three years. The document will describe requirements and timeframes for FDA review of drug development tools and provide a conceptual framework with standards and approaches to support development of biomarkers.
What does this mean for FDA? A Whole Lot of Work!
With the passage of Cures, and with the user fee agreements on the way, FDA staff will have a great deal to do in both the near and long term.
Now that Cures is law, companies, patient groups and other stakeholders will look to FDA for interpretation of the legislative language. FDA will be getting to work almost immediately, if it has not done so already, with deadlines related to key provisions hitting within the first six months to a year, and others spread out over a five- to six-year timeframe.
With the Trump Administration taking the reins in January, some of the Cures implementation flow and cadence will be influenced in ways that will be revealed if and when a new FDA commissioner is nominated.
Also, any industry priorities that did not make it to the finish line with Cures could potentially reappear in the must-pass user fee agreement legislation coming in 2017.
However, what is certain is that FDA will have to execute on a range of activities, including public meetings, new guidance, reports, and other policy documents, over the coming years.
As the folks at FDA wrap up 2016, I know what they will be wishing for in the New Year: more bodies. Since the Cures legislation explicitly allows the Commissioner to make pay scales more competitive and improve the hiring process, hopefully it will allow the agency to more easily recruit and retain top talent to help shoulder the implementation workload.
About the Author:
 Nancy Bradish Myers, president of Catalyst Healthcare Consulting, Inc., is a Washington, DC-based attorney with deep expertise in health care law and regulation, policy development, and government relations. Ms. Myers served as senior policy counsel in the Office of the FDA Commissioner, as well as special assistant / senior strategic advisor to the FDA acting deputy commissioner for operations. She has been closely involved in drug, biotechnology, and medtech regulatory issues for over 25 years. She currently advises select clients on regulatory and health policy matters. You can reach Ms. Myers at nancy@catalysthcc.com, or connect with her on LinkedIn.
Nancy Bradish Myers, president of Catalyst Healthcare Consulting, Inc., is a Washington, DC-based attorney with deep expertise in health care law and regulation, policy development, and government relations. Ms. Myers served as senior policy counsel in the Office of the FDA Commissioner, as well as special assistant / senior strategic advisor to the FDA acting deputy commissioner for operations. She has been closely involved in drug, biotechnology, and medtech regulatory issues for over 25 years. She currently advises select clients on regulatory and health policy matters. You can reach Ms. Myers at nancy@catalysthcc.com, or connect with her on LinkedIn.
Catalyst Healthcare Consulting, Inc. is a strategic policy and regulatory affairs advisory firm. Catalyst partners with biopharma, medical device, combination product, digital health, venture capital, and patient advocacy clients to distill complex health care issues into meaningful commercial and philanthropic opportunities.