
The U.S. Biosimilar Market: Where Is It Today, And Where Is It Going?
By Orestis Mavroudis-Chocholis, Ph.D., Decision Resources Group (DRG)

The U.S. biosimilar market is relatively new. The U.S. government passed legislation to create a regulatory pathway for biosimilars in 2010, with the Biologics Price Competition and Innovation Act (BPCIA), as part of the Affordable Care Act. It took another two years for the FDA to issue the first draft guidelines for developing and registering a biosimilar in the United States, and another three years until the first biosimilar was approved. In September 2015, Sandoz’s Zarxio (filgrastim-sndz) became the first biosimilar to launch in the United States. Since then, the FDA has approved an additional four biosimilars, one of which is on the market (Pfizer’s Inflectra [infliximab-dyyb]), and has issued at least four complete response letters (CRLs). In contrast to the United States, the European Medicines Agency published guidance on biosimilars in 2005, and the first biosimilar (Sandoz’s Omnitrope [somatropin]) was approved and launched in 2006. To date, more than 30 biosimilars have been approved, 26 of which are on currently the market.
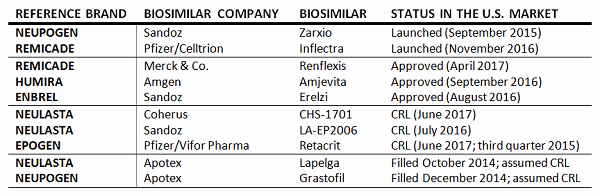
2017 is shaping up to be a pivotal year for the U.S. biosimilar market. Not only the has the FDA issued long-anticipated guidelines on demonstrating interchangeability with a reference product, but the U.S. Supreme Court ruled on the timing of notification for intent to market a biosimilar and involvement in the so-called “patent dance” following the approval of a biosimilar. In addition, the FDA is expected to complete its review of biosimilar applications of top-selling reference brands, including the first trastuzumab and bevacizumab biosimilars.
While the impact of an “interchangeable biologic” label on biosimilar uptake is debatable, with some experts commenting that it does offer a significant commercial advantage and is worth the investment required, others disagree, opining that any advantages are limited and funds can be invested in other areas, such as marketing and educational efforts, to reap equal or even greater rewards. One way or another, biosimilar developers are now aware of what data is required to support an “interchangeable biologic” label, and can more accurately determine the value of such a label and associated investment. The FDA’s Leah Christl, Ph.D., recently commented that interchangeable biosimilars may be two years away from the market, and possibly sooner; such a timeline, however, seems only possible if biosimilar developers have already initiated Phase 3 switching trials that comply with the FDA requirements.
In contrast, the U.S. Supreme Court (SCOTUS) ruling on when biosimilar developers can notify the brand manufacturer of their intent to market a biosimilar has a direct and immediate impact on the U.S. biosimilar market. By ruling that notification can take place before FDA approval, SCOTUS effectively allows for launch of a biosimilar soon after FDA approval, assuming that the 180-day delay post-notification has elapsed, and as such speeds up entry of biosimilars. While this is good news for biosimilar developers, a more significant hurdle currently appears to be the extent of patent litigation post-approval. Even with the patent dance having defined scope and duration, further litigation is possible, and as it is not as restricted by the BPCIA provisions, it can take significantly longer. In the case of Humira, for example, AbbVie is pursuing a patent-litigation defense strategy to delay launch of adalimumab biosimilars, and has stated they can defend patents up to 2022, resulting in a delay of more than five years in the launch of adalimumab biosimilars, given that Amgen’s Amjevita (adalimumab-atto) was approved in September 2016.
Physician Attitudes, Perceptions And Use Of Biosimilars
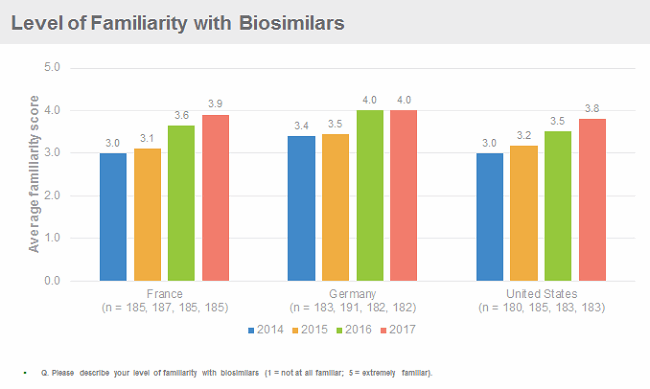
Physicians in the United States are becoming increasingly familiar with biosimilars. Decision Resources Group’s (DRG) research suggests their familiarity is on a par with their European colleagues, even though biosimilars have been available on the U.S. market for almost two years, and only some specialists are able to prescribe them. Importantly, however, U.S. (and European) physicians remain cautious about using biosimilars, citing as the key barriers to uptake concerns about safety, immunogenicity and efficacy, as well as lack of data from clinical trials to support switching. To overcome these barriers, physicians request more data, from bigger clinical trials, from more or all of the indications for which the reference product has been approved, and with longer follow-up, which go above and beyond the regulatory requirements for approval of a biosimilar. If implemented, these requests would make developing a biosimilar a lengthier and more expensive endeavour.
Zarxio and Inflectra have experienced a relatively slow uptake to date in the United States, although Zarxio’s uptake is speeding up. At around one year post-launch, Novartis was reporting Zarxio sales in the United States of more than $100 million, with subsequent reports suggesting that continued and robust Zarxio uptake is a major driver for Sandoz’s revenue growth. By the end of 2016, Zarxio commanded approximately 15 percent of the U.S. filgrastim market, a share that is expected to almost double in 2017, owing to continued uptake of Zarxio and price erosion of Neupogen.
At first glance, Inflectra’s U.S. launch appears to be slower, with Pfizer reporting approximately $25 million in sales in the first four months of launch (vs. $1.2 billion for Remicade in the first quarter of 2017), which could mean that Inflectra’s market share by the end of 2017 will be less than 10 percent, if the market landscape does not change significantly. Importantly, however, Merck & Co. has received FDA approval for Renflexis (infliximab-abda), and may launch in the fourth quarter. As a result of direct competition between Renflexis and Inflectra, both for use and in price, Inflectra’s revenue and market share in 2017 may be even lower.
Dissecting why initial uptake of Zarxio and Inflectra was slow in the U.S. in comparison to the EU5 (France, Germany, Italy, Spain, United Kingdom) is not easy, as multiple factors may be at play, from inclusion of the biosimilars in formularies, ineffective pricing strategies, lack of physician incentives to prescribe biosimilars over the reference brands, and/or physician restraint in prescribing biosimilars. DRG’s research into the launch dynamics of Zarxio and Inflectra in the United States suggests that lack of availability of the drugs was a major constraint. As pricing and availability issues are resolved, biosimilars are added to formularies, payers and providers create stronger incentives for physicians to prescribe biosimilars, and physicians become more comfortable with using them, use is expected to increase significantly over the next two to three years.
How should your biopharmaceutical business adapt its approach when regulations change? Review a case study in this on-demand webinar led by Barry Rosenblatt, CMC expert, to find out.
Biosimilars- Preparing for Opportunities and Challenges
Future Landscape
To date, the FDA has accepted 17 dossiers for review (of the 17, it has approved five and has issued at least four CRLs), but this is only the beginning — 17 additional dossiers are expected to be accepted for review by the end of 2017 alone. In total, 43 biosimilars targeting the U.S. market are in clinical development, with the majority of those in Phase 3 development. Over the past five years, the number of biosimilar R&D projects in the United States has increased significantly, following the definition of the biosimilar regulatory pathway in 2010, issuance of guidance from the FDA since 2012, nearing the time when top-selling branded drugs are coming off patent, and reflects the opportunity for significant revenues for biosimilar developers that enter the U.S. market early. Top brands targeted by biosimilar manufacturers include Humira, Genentech/Roche’s Herceptin and Avastin, and Amgen’s Neulasta, which together represent a market of approximately $20 billion in 2016.
Over the next five years, biosimilars for 12 branded biologics are expected to launch in the United States, including biosimilars of Humira, Rituxan, Avastin, Herceptin, and Neulasta. As a result of continued uptake of filgrastim and infliximab biosimilars, as well as robust uptake of novel biosimilars, the U.S. biosimilar market is expected to grow to approximately $11 billion in 2022, at an annual rate of 57 percent. As a result of biosimilar uptake and brand net cost erosion, the U.S. healthcare system could save a cumulative $50 billion by 2022.
About The Author:
 Orestis Mavroudis-Chocholis, Ph.D., directs a team of analysts who produce Decision Resources Group’s (DRG’s) syndicated Biosimilars solution, providing insights and analysis on the rapidly evolving biosimilar field. DRG’s biosimilar solution focuses on four therapy areas, oncology, immunology, endocrinology, and nephrology, in the major markets (United States, EU5, and Japan). He and the Biosimilars team conduct extensive primary market research with physicians and payers, to uncover the dynamics of the current and future biosimilar market landscape, and secondary market research into the competitive and regulatory landscape. The team also produces interactive forecasts assessing market opportunities of key biologics and their biosimilars and strategically focused content that helps shape biosimilars development and defense strategies.
Orestis Mavroudis-Chocholis, Ph.D., directs a team of analysts who produce Decision Resources Group’s (DRG’s) syndicated Biosimilars solution, providing insights and analysis on the rapidly evolving biosimilar field. DRG’s biosimilar solution focuses on four therapy areas, oncology, immunology, endocrinology, and nephrology, in the major markets (United States, EU5, and Japan). He and the Biosimilars team conduct extensive primary market research with physicians and payers, to uncover the dynamics of the current and future biosimilar market landscape, and secondary market research into the competitive and regulatory landscape. The team also produces interactive forecasts assessing market opportunities of key biologics and their biosimilars and strategically focused content that helps shape biosimilars development and defense strategies.
Prior to his current role, Mavroudis-Chocholis was a director within DRG’s Oncology team, with expertise across several indications including non-small-cell lung cancer, malignant melanoma, and the clinical application of biomarkers in oncology. Prior to joining DRG, he worked at Blueprint Partnership, an independent pharmaceutical market research agency, where he gained extensive expertise in quantitative market research methods. Mavroudis-Chocholis holds a Ph.D. in medicine from the University of Manchester (UK) and a B.Sc. in molecular genetics from the University of Dundee (UK).