
Health-Based Exposure Limits: How Do The EMA's Q&As Compare With New And Forthcoming ASTM Standards?
By Andrew Walsh; Thomas Altmann; Alfredo Canhoto; Ph.D.; Ester Lovsin Barle, Ph.D.; Joel Bercu, Ph.D.; David G. Dolan, Ph.D.; Andreas Flueckiger, M.D.; Igor Gorsky; Jessica Graham, Ph.D.; Robert Kowal; Mariann Neverovitch; Mohammad Ovais; and Osamu Shirokizawa
 This article reviews the European Medicines Agency (EMA) April 16, 2018 Questions and answers on implementation of risk-based prevention of cross-contamination in production and Guideline on setting health-based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities.1 We will discuss the significance of these changes to cleaning validation programs and how they compare to the new ASTM E3106-18 Standard Guide for Science-Based and Risk-Based Cleaning Process Development and Validation2 and the forthcoming ASTM Standard Guide for the Derivation of Health-Based Exposure Limits (HBELs).3 The authors of this article are members of either one or both ASTM standard teams.
This article reviews the European Medicines Agency (EMA) April 16, 2018 Questions and answers on implementation of risk-based prevention of cross-contamination in production and Guideline on setting health-based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities.1 We will discuss the significance of these changes to cleaning validation programs and how they compare to the new ASTM E3106-18 Standard Guide for Science-Based and Risk-Based Cleaning Process Development and Validation2 and the forthcoming ASTM Standard Guide for the Derivation of Health-Based Exposure Limits (HBELs).3 The authors of this article are members of either one or both ASTM standard teams.
Table 1 shows a comparison of the original questions and answers in the EMA’s draft document versus the modified questions and answers.
Table 1
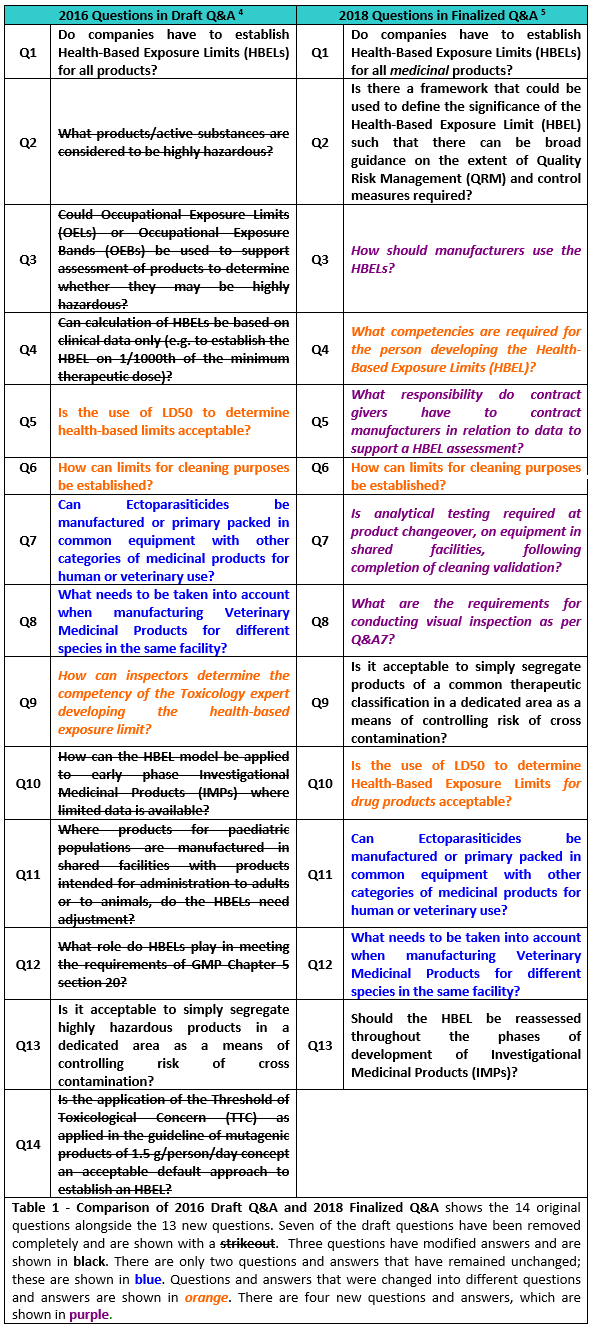
The immediate impression readers will get from Table 1 is that the draft questions and answers (Q&As) underwent significant changes. One of the issues with the draft Q&As was that there was no public solicitation by the EMA of pharma industry companies and organizations in the form of working groups prior to publishing the draft Q&As. Consequently, the draft Q&As came as a surprise to most of the industry. Many of the answers were seen as problematic, as they were directing the industry away from the science- and risk-based approach that regulators have been promoting since 2001. In addition, the draft Q&As contradicted the 2012 Concept Paper6 and reverted to traditional approaches that have been demonstrated to need replacement with modern science- and risk-based approaches.7-12 As a result, 32 industry organizations, companies, and individuals, including the ASTM E3106 team, submitted extensive comments on the draft Q&As. These comments were significant enough for the EMA to hold a workshop on June 20-21, 2017 to discuss the comments. Subsequently, in its final Q&A document, posted on its website on April 16, 2018, the EMA introduced numerous changes.
The full text of each of the Q&As and their updates are shown in the boxed areas and are discussed one by one.
Q1 (2016 Draft) − Do companies have to establish Health-Based Exposure Limits (HBELs) for all products?
A: Yes, HBELs should be established for all products. HBELs for highly hazardous products are expected to be completed in full as per the EMA guide (EMA/CHMP/CVMP/SWP/169430/2012) or equivalent. See Q2 for products/active substances considered to be highly hazardous. Products that do not fall into the highly hazardous category may be addressed as per Q4.
Q1 (2018 Final) − Do companies have to establish Health-Based Exposure Limits (HBELs) for all medicinal (emphasis added) products?
A: Yes, HBELs should be established for all medicinal products.
The toxicological or pharmacological data, on which the HBEL calculation relies, requires periodical re-assessment throughout a product’s life cycle.
Discussion: There are two important changes to Q1. The first is that the establishment of HBELs has been narrowed to "medicinal" products. This apparently clarifies that not all chemicals used in manufacturing by a pharmaceutical company may be medicinal. This was unnecessary, as EMA's November 2014 Guidance1 already stated that it applied to medicinal products. What is not obvious is that this now can be interpreted to exclude compounds that are intermediates, degradants, and cleaning agents, for example. This is a concern since intermediates, degradants, and cleaning agents are all possible hazards for cross contamination of another product. This indirectly ties into the changes in Q5 (now Q10), where the LD50 is only excluded from calculating HBELs in drug products, possibly indicating that its use is acceptable for other compounds used in drug production. This is a serious concern, as the historical use of the LD50 divided by an arbitrary adjustment factor for calculating cleaning limits for cleaning agents has been shown to be very unreliable and significantly overly conservative. This is due to the large composite adjustment factors historically and unjustifiably used to extrapolate from an LD50 (and endpoint of mortality) to a human safe dose.9, 13 In most cases, these LD50-based limits for cleaning agents are so conservative that some companies have felt forced to stop using cleaning agents completely. Furthermore, the LD50 limit is not convertible or relatable to any meaningful toxicological hazard assessment that could be used to derive health-based limits; therefore, this needs to change.
On the positive side, Q1 now makes no reference to "highly hazardous" compounds or to Q&A2, which referred to a list of criteria that would indicate if the substance should be considered as such. The term "highly hazardous" was a major source of objections from many organizations, including the ASTM Cleaning Standard Team, since this proposed two-level classification did not consider the general toxicological principle that “the dose makes the poison,” nor did it consider the ease or difficulty of cleaning or degrading a compound. Today, it should be understood that the hazards associated with all drugs are associated with the doses at which these hazards appear. For example, two drugs may have the same intrinsic hazards, but the effects can appear at doses that may be several orders of magnitude apart. It is vital that each drug is individually addressed based on its hazard and dose-response and drugs should not be classified into simplistic categories. This would be contrary to the guidance already provided in the Risk-MaPP (Risk-based Manufacture of Pharmaceutical Products) Guideline in 2010,14 as well as the ASTM E3106-18 standard in 2017.2
Q2. (2016 Draft) − What products/active substances are considered to be highly hazardous?
A: Highly hazardous products are those that can cause serious adverse effects at low doses and that therefore would benefit from a full toxicological assessment in order to derive a safe HBEL.
Highly hazardous products are identified based on their inherent toxicological and pharmacological characteristics and include the groups below (this list is not an exhaustive list and if evidence is available indicating that the product may cause adverse effects at low doses by other mechanisms it should be considered as highly hazardous).
Manufacturers should consider, via a safety assessment against the guidance below, if products/active substances should be considered highly hazardous. Evidence indicating a product or active substance falls within any of the categories below should result in a product being considered highly hazardous. If in doubt, manufacturers should consider the product potentially highly hazardous and apply the EMA guide (EMA/CHMP/CVMP/SWP/169430/2012) in full to derive a safe HBEL.
1. Genotoxic (specifically mutagenic) compounds that are known to be, or highly likely to be, carcinogenic to humans. Compounds of this group are easily identifiable, since genotoxicity would be related to the pharmacology, e.g., as DNA alkylating cytostatics, and their use is usually restricted to oncology indications with respective warning statements in the Summary of Product Characteristics.
2. Compounds that can produce reproductive and/or developmental effects at low dosages, for example where evidence exists of such effects being caused by a clinical dose of <10 mg/day (veterinary dose equivalent 0.2 mg/kg/day) or dosages in animal studies of ≤1 mg/kg/day.
3. Compounds that can produce serious target organ toxicity or other significant adverse effects at low doses, for example where evidence exists of such effects being caused by a clinical dose of <10 mg/day (veterinary dose equivalent 0.2 mg/kg/day) or dosages in animal studies of ≤1 mg/kg/day.
4. Compounds with a high pharmacological potency, i.e., recommended daily dose of <1 mg (veterinary dose equivalent 0.02 mg/kg).
5. Compounds with a high sensitising potential.
Q2 (2018 Final) − changed to: Is there a framework that could be used to define the significance of the Health-Based Exposure Limit (HBEL) such that there can be broad guidance on the extent of quality risk management (QRM) and control measures required?
A: Firstly, it should be recognized that hazard varies on a continuum scale and that there are no firm cut off points, risk should be controlled on a proportionate basis. However, as a broad hypothetical model, the following figure could be considered to show the increasing level of hazard (red being highest hazard) presented by products and there should be a commensurate increase in the level of control to prevent potential cross contamination in a shared facility. Actual HBEL values should be used in QRM studies to determine the actual controls required.

Figure 1: Diagram developed from an original concept published by ISPE. Source: ISPE Baseline® Pharmaceutical Engineering Guide, Volume 7 – Risk-Based Manufacture of Pharmaceutical Products, International Society for Pharmaceutical Engineering (ISPE), Second Edition, July 2017.
Discussion: Q2 underwent the most extensive changes of all the Q&As and has dropped the original “highly hazardous” classification and categorization approach.
While this change represents a significant advancement to the continuum of hazards philosophy as shown in Figure 1, a more accurate and more useful scale for visualizing and comparing HBELs has already been developed and can be used as an improved reference for this purpose.15 This new scale is continuous, as it is based on actual HBEL data and, as it was developed for potential use in cleaning failure modes and effects analyses (FMEAs), it has the familiar 0 to 10 range. The scale has also been shown to contain the HBELs of the majority of pharmaceutical compounds. Since the values on this scale are derived directly from the established HBELs, this provides a manufacturer or a regulator a simple tool to quickly visualize the relative hazard of a compound. This scale can be adopted by companies and regulators for their QRM procedures for both cleaning and containment. Figure 2 provides details regarding the new scale.

Readers may find it surprising to see arsenic trioxide, a compound most people would consider to be quite hazardous, occupying the midpoint of the scale. This is the ADE concept at work: The toxicity of a compound is dose-dependent, so while higher doses of a compound may generate extreme adverse effects, somewhat lower doses may be harmless. The Toxicity Scale helps to accurately reveal this in relation to other compounds/APIs. We believe the use of this scale is preferable to using a simplistic "Highly Hazardous/Non-Hazardous" approach to evaluating the hazard posed by a residual level of a compound.
Q3. (2016 Draft) − Could Occupational Exposure Limits (OELs) or Occupational Exposure Bands (OEBs) be used to support assessment of products to determine whether they may be highly hazardous?
A: Yes. Extrapolation of an OEL or OEB (lower end of the range) to a preliminary Permitted Daily Exposure (PDE) can be simply done by using the following formula: PDE (μg/day) = OEL (μg/m3) x 10 m3 (the volume air breathed by a worker in 8 hours). Additional adjustment factors may be needed due to potential differences in target population (worker vs. patient), route of exposure, etc. If the resulting PDE value is 10 μg/day or lower, the product should be considered as highly hazardous.
Q3. (2018 Final) − This Q&A was eliminated and changed to a different question
Discussion: Presumably, based on the many serious objections by many organizations and companies, including the ASTM Cleaning Standard Team, regarding using OELs or OEBs, Q3 was eliminated completely. While the simple conversion of an OEL to a provisional HBEL can be done, the point of departure and the study used for an OEL may not be applicable for HBELs for other routes of exposure. In addition, adjustment factors may need to be changed due to potential differences in the target population (worker vs. patient), the route and the duration of exposure, etc. Rather than manipulating an OEL calculation, it is more sensible to perform the correct HBEL derivation. OEBs should not be used due to their highly variable default values and inconsistent practice from company to company. Since the effective date of the EMA guideline, some companies may have used existing OELs and OEBs for estimating the HBELs; however, the majority of companies simply moved forward by providing a full toxicological assessment with a focus on the patient safety. Now, nearly four years after publication, these approaches are unnecessary. Instead of expending their time and resources justifying the conversion of OELs and OEBs into provisional HBELs, companies should just simply proceed with developing appropriately derived HBELs.
Q4. (2016 Draft) − Can calculation of HBELs be based on clinical data only (e.g., to establish the HBEL on 1/1000th of the minimum therapeutic dose)?
A: Many existing commercial products and new products for which clinical safety profiles are well established and that do not belong to the highly hazardous category (see response to Q2) have a favourable therapeutic index (also referred to as the therapeutic window or safety window). This means that unwanted or adverse health effects (that may have been identified as toxic effects in animal studies at high doses) may occur - if at all - at dose levels orders of magnitude above the therapeutic dose range and the pharmacological activity would therefore be the most sensitive/critical effect. In this situation, therapeutic dose information could be used as the ‘Point of Departure’ for calculation of an HBEL (e.g., the PDE). Under these circumstances, HBEL based on the 1/1000th minimum therapeutic dose approach would be considered as sufficiently conservative and could be utilized for risk assessment and cleaning purposes.
Q4. (2018 Final) − This Q&A was eliminated and changed to a different question.
Discussion: Thankfully, Q4 was eliminated. This answer contained two major problems. First, it incorporated the draft categorization of drugs into "highly hazardous" and "non-hazardous drugs." Second, it perpetuated the continued use of an arbitrary assessment factor of 1/1,000th of the minimum therapeutic dose approach as an appropriate means for establishing HBELs for "non-hazardous" drugs. As mentioned above, the use of an arbitrary assessment factor, in particular 1/1,000th of a dose, is very unreliable and may significantly over- or underestimate a chronic dose that is safe for humans. Thus, it may have been underprotective for patients administered a residual carryover of some drugs and overprotective for other drugs, resulting in an unnecessary burden to low-hazard and low-risk product manufacturers prior to the adoption of HBELs.7-12 Nonetheless, while Q4 has been eliminated, use of an arbitrary adjustment factor of 1/1,000th still persists in the form of "historical limits" in Q6. Furthermore, the continued use of the 1/1000th of a dose limit contradicted EMA’s previous Concept Paper.6
A previously published article described cases where the 1/1,000th of the minimum therapeutic dose approach is not adequately protective.12 The benefit of an HBEL, also known as a permitted daily exposure (PDE) and acceptable daily exposure (ADE), is that it is calculated by an expert in a form of a daily dose. Conversely, the identification of minimum therapeutic doses and their extrapolation to a safe dose has typically been performed by cleaning validation personnel and did not involve an evaluation by an expert in a toxicological risk assessment. Again, dividing the minimum therapeutic dose by a factor of 1,000 for a drug with only minor hazards is overly conservative and has imposed unnecessary burdens upon low-risk drug manufacturers. The upcoming ASTM Standard for the Derivation of Health-Based Exposure Limits (HBELs) will provide guidance from expert pharmaceutical toxicologists on how to handle setting limits for low-risk drugs.
Q5. (2016 Draft) − Is the use of LD50 to determine health-based limits acceptable?
A: No, LD50 is not an adequate point of departure to determine an HBEL.
Changed to Q10 (2018 Final). Is the use of LD50 to determine Health-Based Exposure Limits for drug products acceptable?
A: No, LD50 is not an adequate point of departure to determine a HBEL for drug products.
Discussion: The change in Q5 is subtle, and the EMA may have been convinced it is a minor point to clarify that it only applies to drug products. However, the original answer was correct, and the LD50 should never be used to calculate an HBEL for any compound and, in particular for cleaning validation, for the components of cleaning agents. Members of the ASTM Cleaning Standard Team published an article in 2013 demonstrating why the LD50 cannot be used as a point of departure to derive a NOEL (no observable effects level) and how the historical LD50 approaches being used to calculate limits for cleaning agents are highly inaccurate and lead to excessively low limits. In many cases, companies have been unable to reliably achieve these limits analytically. 12 The use of the LD50 has led to the pharmaceutical industry moving away from or even ceasing to use, cleaning agents, which appears to be completely unjustified, as many cleaning agent components are quite innocuous and safe. The elimination of cleaning agents can lead to inefficient cleaning cycles and may have even led to product residues remaining on manufacturing surfaces in some cases.
The use of the LD50 is not endorsed scientifically for calculation of any HBELs. LD50s are the expression of single-dose (acute) toxicity, but HBELs, by definition, must be set for long-term (chronic) exposure. An attempt to statistically correlate known LD50 values (acute toxicity) with their corresponding NOELs (chronic toxicity) was so poor that it was shown that the LD50 cannot be used to set a limit of chronic toxicity.9 For drugs, there is no need to use an LD50 to establish an HBEL, since much more relevant chronic toxicity data exists. Also, oral LD50 testing is seldom performed for small molecule drugs and is of no value for development of HBELs for parenterally-administered follow-on drugs. The selection of an appropriate point of departure (PoD) for HBEL calculations of commercially available chemicals, or pharmaceutical intermediates that do not have full toxicological datasets, can be a challenge, but does not warrant the use of the LD50. Alternative and scientifically acceptable recommendations are described in the upcoming ASTM Standard Guide for the Derivation of Health-Based Exposure Limits (HBELs) to provide guidance on this issue.
Q6 (2016 Draft & 2018 Final) − How can limits for cleaning purposes be established?
A: Although the EMA guideline (EMA/CHMP/CVMP/SWP/169430/2012) may be used to justify cleaning limits (as per Introduction paragraph 3), it is not intended to be used to set cleaning limits at the level of the calculated HBEL (using the guideline methodology). The cleaning limits should continue to be based via risk assessment and additional safety margins to help account for uncertainty in the cleaning processes and analytical variability. Traditional cleaning limits used by industry such as 1/1,000th of minimum therapeutic dose or 10 ppm of one product in another product, may accomplish this for non- highly hazardous products. For products classed as highly hazardous, where a thorough risk assessment can justify manufacture in shared facilities, cleaning limits should include safety factors beyond the HBEL and should not be higher than the traditional cleaning limits approach.
New answer
A: Although the EMA guideline (EMA/CHMP/CVMP/SWP/169430/2012) may be used to justify cleaning limits (as per Introduction paragraph 3), it is not intended to be used to set cleaning limits at the level of the calculated HBEL.
For existing products, manufacturer’s historically used cleaning limits should be retained and can be considered alert limits provided that when taking cleaning process capability into account, they provide sufficient assurance that excursions above the HBEL will be prevented. A similar process should be adopted when establishing cleaning alert levels for products introduced into a facility for the first -time.
Results above the alert cleaning limit should trigger an investigation and, where appropriate, corrective action to bring the cleaning process performance within the alert cleaning limits. Repeated excursions above the alert cleaning limit will not be considered acceptable where these indicate that the cleaning method is not in control. Recognized appropriate statistical methods may be used to determine whether the cleaning process is in control or not.
Discussion: Q6 is the most significant of the Q&As regarding cleaning validation, and EMA is to be commended for adopting its emphasis on a statistical approach; however, we have some suggestions for improvement. While the changes to this answer are an improvement on the original answer, the new answer continues several misunderstandings of the principles of science and risk involved with setting cleaning limits that the ASTM E3106-18 standard provides. The new answer can be misinterpreted, and some companies/practitioners are claiming that this answer allows the old limits to stay in place and they have no plans to make any changes to their cleaning programs or toward increasing understanding of their cleaning processes. That is a serious misconception if one understands the context around this Q&A and reads carefully what is being stated.
The first sentence remains the same and correctly reflects the understanding that HBELs should be used for assessing the risks involved in cleaning but should not be used for setting the day-to-day control limits for use in cleaning validations. Any control limits used for cleaning should be set based on cleaning data using statistical process control. The reasons why HBELs should not be used for setting cleaning limits, and should only be used for risk assessment, were explained in 2011 in a two-part article,7,8 and these are now embodied in the ASTM E3106-18 standard. 2 Basically, while the safe residue levels calculated from HBELs must be safe regarding patient exposure, they may be well above the level of residues easily achievable in cleaning, even easily visible, and therefore are inappropriate to use as cleaning limits. These same reasons apply in many cases to the 1/1,000th and 10 ppm limits as well. Consequently, a different approach is necessary for setting control limits for these compounds in either case.
The second paragraph states that "historical" limits should be retained as alert limits, provided they can assure data will not exceed the HBELs. On the basis of the following analysis, the ASTM teams do not agree with its practice. Afterward, we present our proposed solution.
In a study of 304 pharmaceutical compounds, it was found that HBELs were absolutely necessary for determining the safe residue levels for evaluating cleaning validations.12 For about 15 percent of these compounds, the limits based on 1/1,000 dose criterion were found to be not safe enough. However, for the remaining 85 percent of the compounds, the safe residue levels calculated from HBELs start to rise well above the residue levels easily achievable for these compounds and therefore are unsuitable for use in controlling cleaning processes.
Figure 3 shows calculated total organic carbon (TOC) swab limits for these 304 compounds, both for their PDEs and their corresponding historical limits. Hypothetical TOC swab data has been superimposed on these limits at the 100 ppb level to evaluate how the data compares to both limits. 100 ppb was chosen because it is representative of good cleaning, but typical TOC results can be above or below this level. As described in the Figure 3 caption, there are very few instances where the historical limits might be used as alert limits for cleaning processes. If TOC swab results begin to approach 1 ppm (1,000 ppb), fewer and fewer historical limits are useable, even though the process capability for the corresponding PDE may be excellent.

Figure 3: Graph of TOC swab limits calculated for the 304 compounds for both PDE-based limits and the 1/1,000th dose or 10 ppm-based limits (note: these TOC limits are plotted on a logarithmic scale). A hypothetical run of TOC swab results has been superimposed on the graph at the 100 ppb level. As we see in region ①, the 1/1,000th limits are below the TOC swab data, so TOC cannot be used at all. There are even some compounds (region ②) that defaulted to the 10 ppm limit, but the PDE limits are below these. In region ③ there are many compounds where the 1/1,000 limits are well above the TOC data and their use as alert limits is debatable. In region ④ nearly all of the historical limits (both the 1/1000th and 10 ppm) are too far above the TOC data to be used as alert limits. (Note: the 10 ppm limit is set on the level of the batch and not as an analytical limit. The TOC limit for TOC being 10 ppm is coincidental.) Graph created in R by Mohammad Ovais
The third paragraph then discusses how to use these "historical" limits as alert limits to initiate investigation and corrective actions for excursions or indicate whether a cleaning process is not in control. The following examples will show how the use of many historical limits as alert limits will not provide appropriate warning of cleaning process issues.
TOC swab data from eight cleaning validation runs were obtained from a recent successful cleaning validation study and used for this analysis. This is very good cleaning data with very low TOC residues found on the equipment surfaces. An Xbar Control Chart of the data was created using Minitab (Figure 4). An Xbar Control Chart is used to study how a process changes over time using the averages of data plotted in sequence. The Xbar Chart in Figure 4 plots the average of seven TOC data points obtained for each cleaning run. An Xbar control chart always has a central line derived from the average of the sample averages, an upper line for the upper control limit (average +3 standard deviations), and a lower line for the lower control limit (average -3 standard deviations).

Figure 4: Minitab Xbar Chart of TOC swab data for eight cleaning runs (seven swab results each) showing that the data averaged 85 ppb of TOC. Minitab calculated an upper control limit (UCL = 132) from the average and standard deviation of this data set. (Note: while Minitab also calculates a lower control limit, with cleaning data only a UCL is needed.)
Xbar Control Charts are one of the basic tools of statistical process control (SPC) and have been used to set statistical limits for controlling processes based on data collected from the process itself. The control chart was first described by Dr. Walter Shewhart nearly 100 years ago17 and has since been used across nearly every industry, with applications wherever data is collected. The new ASTM E3106-18 standard promotes the use of SPC for setting cleaning limits based on data derived from the cleaning process. ASTM E3106-18 states that data that has been collected in the risk evaluation stage can have SPC limits calculated from this data and that these SPC limits should then be used for monitoring cleaning processes. This use of SPC is aligned with the FDA's Process Validation Guidance, where it states that in-process specifications "...shall be derived from previous acceptable process average and process variability estimates where possible and determined by the application of suitable statistical procedures where appropriate." The FDA's Process Validation Guidance also expects manufacturers to understand the sources of variation, detect the presence and degree of variation, understand the impact of variation on the process, and control the variation in a manner commensurate with the risk it represents to the process. Accordingly, the use of SPC with cleaning data will allow manufacturers to detect, understand, and control variations in cleaning processes in a manner commensurate with the risk. Applying SPC to cleaning data was first suggested and described in 20118 and is now included in the ASTM E3106-18 Standard Guide under the section on Risk Control. As discussed in a previous article, this can even be applied to HPLC data, where much data may be below detection limits.18
Our recommended solution is to establish cleaning limits based on SPC techniques. In Figure 5 we see statistical process control limits derived from the cleaning data in Figure 4 being used as control limits, with two new hypothetical cleaning runs, nine and 10, added. Clearly the data for these runs is significantly different from the cleaning data of the previous eight runs and should be immediately investigated and brought back under control. Considering that almost all HBELs would be much higher than the data shown here, there would probably be no risk to patient safety and no product would have been compromised. Regardless, the deviation of the data from the previously experienced data is very significant and should be investigated.

Figure 5: Minitab Xbar Chart of TOC swab data showing the upper control limit (UCL) only from the previous eight runs. The averages for runs nine and 10 are significantly higher than the previous eight runs, and Minitab has flagged them as red points. Note: The LCL was removed from this graph, as lower limits are not appropriate for cleaning data.
As the third paragraph suggests, historical limits can be used as alert limits, but this is not reasonable or scientifically justifiable. To illustrate why historical limits are inappropriate as alert limits, six compounds from across the spectrum of the 304 compounds shown in Figure 1 were selected from the four regions shown and swab limits for TOC were calculated for them. In Figure 6 we see the historical limits for these six compounds shown as alert limits for the data in this example.

Figure 6: Evaluation of the use of historical limits for six compounds as alert limits for monitoring cleaning data. Note: Lower control limit (LCL) was not removed from this graph.
Why are these historical limits not useful as alert limits? Even the lowest of these historical limits (Basimglurant) is more than 25X higher than the UCL (132 ppb). The others are far worse. Although the residue data is climbing significantly above the previously obtained residue data, none of these limits will raise an alert, as they are all too far above the data.
Compounding this problem even further is the fact that all historical limits are calculated from several parameters (maximum daily doses, total surface areas, etc.) that can vary significantly from product to product, manufacturing train to manufacturing train, facility to facility, and company to company. In some cases, they will go down and in some they will go up even more. There will never be any consistency to alert limits based on historical limits and, what we believe is worse, historical limits do not put any focus on the cleaning process and keeping it under control.
It is likely that the inclusion of these historical limits by the EMA was a compromise with some companies and individuals who have objected to the movement to HBELs and want to keep the historical approaches in place without providing a scientific, risk-based rationale. As mentioned above, the HBEL-based limits and the historical limits are both inappropriate for setting cleaning limits. Instead, limits should be set using SPC techniques after data has been collected during qualification of the cleaning process and the risk has been shown to be acceptable by evaluating the collected data against HBEL-based limits using process capability.
We hope that the final sentence in Q&A6 will get the industry moving in the right direction for the future of cleaning validation in that "recognized appropriate statistical methods" will eventually become required "to determine whether the cleaning process is in control or not." Once it is realized that the historical limits provide no real assurance of control, their continued use should be rapidly abandoned.
We hope this discussion helps to explain why Q&A6 is still problematic and should be modified again in the very near future to remove any references to the non-scientifically based 1/1000th of a dose or 10 ppm in the next batch limits.
Q7 (2016 Draft) − Can Ectoparasiticides be manufactured or primary packed in common equipment with other categories of medicinal products for human or veterinary use? [Now Q11 in the 2018 final Q&A document. See discussion below]
A: If HBEL data cannot support manufacture in shared facilities then the Ectoparasiticides should be manufactured in dedicated facilities.
Changed to Q11. Can Ectoparasiticides be manufactured or primary packed in common equipment with other categories of medicinal products for human or veterinary use?
A: If a HBEL cannot be determined or data cannot support manufacture in shared facilities then the Ectoparasiticides should be manufactured in dedicated facilities.
Discussion: The only change to this Q&A is the addition of the exception "If a HBEL cannot be determined...."
Q8 (2016 Draft) − What needs to be taken into account when manufacturing Veterinary Medicinal Products for different species in the same facility?
A: The guideline on setting health- based exposure limits indicates that the carry over limit should generally be derived using the human PDE. However, in cases where there is particular concern relating to known sensitivity of a particular species (e.g., Monensin in horses) a Health-Based Exposure Limit (HBEL) approach taking into account specific animal toxicity knowledge should be used. For non-highly hazardous products, the approach described in the response to question 6 can also be applied.
Changed to Q12 (2018 Final) − What needs to be taken into account when manufacturing Veterinary Medicinal Products for different species in the same facility?
A: The guideline on setting health-based exposure limits indicates that the carry over limit should generally be derived using the human HBEL.
However, in cases where there is concern relating to known susceptibility of a particular species (e.g., monensin in horses), the HBEL approach should take into account knowledge of specific animal toxicity when evaluating products manufactured in shared facilities/equipment.
Discussion: The change to this Q&A eliminates the last sentence in the draft, removing the reference to non-highly hazardous products.
Q9 (2016 Draft) − How can inspectors determine the competency of the Toxicology expert developing the health-based exposure limit?
A: Inspectors should evaluate the company's assessment of the competence of their expert in the field by reviewing justification of experience and qualification.
Changed to Q4 (2018 Final) − What competencies are required for the person developing the Health-Based Exposure Limits (HBEL)?
A. Health-Based Exposure Limits should be determined by a person who has adequate expertise and experience in toxicology/pharmacology, familiarity with pharmaceuticals as well as experience in the determination of health-based exposure limits such as Occupational Exposure Levels (OELs) or Permitted Daily Exposure (PDE).
Where experts are contracted to provide the HBEL, contractual agreements in compliance with Chapter 7 requirements should be in place prior to work being conducted. It is not considered acceptable for manufacturers to ‘purchase’ HBEL assessments without recording an assessment of the suitability of the provider (including the specific technical expert) as a qualified contractor.
Discussion: HBELs must be prepared by individuals with specific education and training in toxicology/pharmacology/pharmacotherapy and risk assessment methods who can apply the principles of toxicology and pharmacology to deriving a safe limit. An ASTM Standard Guide for Derivation of Health Based Exposure Limits (HBELs) is currently in development and will provide guidance.3 A curriculum vitae (CV) should be available on request that demonstrates the educational background (e.g., toxicology, pharmacology, medical, or other related disciplines), any certifications such as the Diplomate of the American Board of Toxicology (DABT) or European Registered Toxicologist (ERT), the years of experience in the field, and any publications related to the field. While all of these are not required for a “qualified expert,” the appropriate documentation in these areas demonstrates the expertise to work in this area.
Q10. How can the HBEL model be applied to early phase Investigational Medicinal Products (IMPs) where limited data is available?
A: Health- based exposure limits should be established based on all available data and as such assessments associated with IMPs should be regularly reviewed for presence of new data. Toxicology experts should also make judgments about the future potential of the material to demonstrate critical effects where key toxicological testing has not been completed (e.g., this may be based on comparison to other similar molecules where available) and any additional adjustment factors that may be appropriate. This would allow manufacturers to assume worst case and make sound judgments on the level of organizational and technical control measures required.
Changed to Q13. Should the HBEL be re-assessed throughout the phases of development of Investigational Medicinal Products (IMPs)?
A: Health-Based Exposure Limits should be established based on all available data, and particularly as the knowledge base for IMPs is continually evolving the basis for establishing the HBEL, should be regularly reviewed taking account of any new relevant data.
Discussion: The change to Q&A10 dropped the requirement that "toxicology experts" predict what HBELs should be without data in hand and what adjustment factors should be applied.
Q12 (2016 Draft) − What role do HBELs play in meeting the requirements of GMP Chapter 5 section 20?
A: Once the health- based assessment has been completed and HBEL confirmed, these data should be used via a Quality Risk Management process to assess if current organisational and technical control measures are adequate, or in the case of new equipment/facility to determine what control measures are required. It is expected that the higher the hazard of products/active substances, the higher the inherent risk and the more significant organisational and technical control measures will be required. Health based exposure limits provide an accepted safe level of cross contamination and they should be used to justify cleaning limits.
Q12 (2018 Final) - This Q&A was eliminated and changed to a different question.
Discussion: The draft Q12 was eliminated.
Q13 (2106 Draft) - Is it acceptable to simply segregate highly hazardous products in a dedicated area as a means of controlling risk of cross contamination?
A: Manufacturers cannot just segregate highly hazardous products from other lower risk products as a means of dealing with the risk to patient safety. This may protect less hazardous products from contamination but it does not address the possibility for cross contamination between highly hazardous products. The approach taken to address cross contamination between individual highly hazardous products produced in the same dedicated area should be justified, taking account of the clinical application and toxicological profile of the individual products within the group of products manufactured in the dedicated area. This should include implementation of appropriate technical and organisational control measures.
Changed to Q9 (2018 Final) − Is it acceptable to simply segregate products of a common therapeutic classification in a dedicated area as a means of controlling risk of cross contamination?
A: Manufacturers cannot just segregate common products from other product types as a means of dealing with the risk to patient and animal safety. Although this may prevent contamination of other product classes, it does not address the possibility for cross contamination within product classes. The approach taken to control cross contamination between individual products within a class produced in the same dedicated area should follow the principles in Q&A 3. This should include implementation of appropriate organisational and technical control measures to prevent contamination between such products within product specific HBELs.
Discussion: The draft Q13 was moved to Q9. The modifications may appear minor but are very significant.
The question was changed from segregating "highly hazardous products" to segregating "products of a common therapeutic classification." This may raise the question in the industry of whether or not EMA considers that equipment dedication should fall along common therapeutic classes.
The first sentence dropped the term "highly hazardous products" and "lower risk products" and replaced them with the term "common products," which is not defined. This sentence was also expanded to include animals. The second sentence eliminated the distinction between "less hazardous" and "highly hazardous" products and referred to "product classes," but this new term is also undefined. It is not clear whether the EMA may consider products that share common equipment to be in a particular "product class" or is expecting companies to define classes of products that can share common equipment. The third sentence was modified to remove "... should be justified taking account of the clinical application and toxicological profile of the individual products within the group of products manufactured in the dedicated area" and replace it with "... should follow the principles in Q&A3." As stated above in the discussion of Q&A3, we are relieved to see a return to a risk-based approach. Finally, the phrase "... to prevent contamination between such products within product specific HBELs" was added to the last sentence.
While these modifications are improvements on the original answer, it is our opinion that the possibility of sharing equipment should be decided solely by the level of risk presented and the ability of the company to control or mitigate that risk, not by a shared therapeutic action or other type of classification. Conversely, the requirement to dedicate any product should be decided solely based on the level of risk being unacceptable and/or the inability of the company to mitigate or control the risk presented.
Q13 (2018 Final) − Should the HBEL be re-assessed throughout the phases of development of Investigational Medicinal Products (IMPs)?
A: Health-Based Exposure Limits should be established based on all available data, and particularly as the knowledge base for IMPs is continually evolving, the basis for establishing the HBEL, should be regularly reviewed, taking account of any new relevant data.
Discussion: The HBEL should be reassessed periodically when significant data emerges that may affect the HBEL calculation and/or its assumptions. At a minimum, the IMP HBELs should be assessed once there is an adequate dataset (repeat-dose animal studies, mutagenicity information, PK/PD data, FIH data) and reevaluated prior to marketing when there is additional clinical experience. Ideally, the HBEL is also periodically reviewed as new relevant clinical data and longer nonclinical study data emerges. The HBEL review should become a part of the cleaning validation risk management program to increase manufacturers' maturity levels in that area.
Q14 (2016 Draft) − Is the application of the Threshold of Toxicological Concern (TTC) as applied in the guideline of mutagenic products of 1.5 μg/person/day concept an acceptable default approach to establish an HBEL?
A: Yes, except in the case of highly sensitising active substances and products.
Discussion: Q14 was eliminated. However, it should be noted that in the case of a compound with a limited dataset, where the material may pose a mutagenicity hazard as the critical effect, the application of the TTC of 1.5 µg/person/day could be applied as an interim limit until additional information is available to aid in the calculation of a proper HBEL.
The NEW Q&As:
Q3. How should manufacturers use the HBELs?
A: The role of HBELs in determining cleaning limits is explained in Q&A 6. However, the purpose of generating HBELs goes beyond justification of cleaning limits.
Once the health-based assessment has been completed and the HBEL confirmed, these data should be used via a Quality Risk Management process to determine what controls need to be put in place and to assess if existing organisational and technical control measures are adequate or if they need to be supplemented. This Quality Risk Management process should be carried out prospectively in the case of new equipment/facility to determine what control measures are required.
It is expected that for products which present a higher potential harm to patients/animals, more elaborate organisational and technical control measures will be required. Using a structured Quality Risk Management process, manufacturers should consider the risks of cross contamination down to the established level from the HBEL. During the QRM study manufacturers should consider how easily such a quantity of contamination could occur, without detection, at batch and unit dose level.
The level of detail in the QRM process should be commensurate with the potential harm as indicated by the HBEL and the suitability of control measures supported by practical and science-based evidence.
Manufacturers should be mindful that cross contamination controls implemented previously may not adequately assure control of the cross-contamination risk in the context of the HBEL approach.
Additional observation of working practices, investigation, and analysis may be required to provide full practical confidence in the effectiveness of controls.
Where control measures cannot adequately assure that the potential contamination is consistently controlled to a level below that of the HBEL, then the products concerned should be manufactured in dedicated facilities.
Discussion: This new Q3 is a very welcome return to the application of science and risk to cleaning that had its beginnings in the Risk-MaPP Guide and is now formalized in the new ASTM E3106-18 standard. The ASTM E3106-18 science- and risk-based approach as applied to cleaning has been described in more detail in the article Measuring Risk in Cleaning: Cleaning FMEA and the Cleaning Risk Dashboard.16 That article describes how the toxicity scale for HBELs can be combined with a process capability scale derived from data collected during cleaning to quantify the risk involved in a particular cleaning process so that appropriate control measures can be put in place. The use of detectability scales for the appropriate selection of analytical methods, including visual inspection, is also described. This article provides more detail than is contained in the ASTM E3106-18 standard, along with a case study, on how to use the HBEL and implement a science- and risk-based approach that is completely consistent with EMA's new guidance in Q3.
New Q5 (2018 Final) − What responsibility do contract givers have to contract manufacturers in relation to data to support a HBEL assessment?
A: Companies engaging a contract manufacturer to make a drug are obliged to either provide an HBEL, or to provide all available nonclinical and clinical data to the contract manufacturer for them to either calculate the HBEL itself, or to outsource the calculation of an HBEL to a qualified consulting toxicologist.
Discussion: This new Q5 is a welcome clarification that it is the responsibility of the contract giver/contracting company to provide appropriate quality support to the contract acceptor/contracted company, which includes the provision of HBELs or data to support their derivation.
Q7. Is analytical testing required at product changeover, on equipment in shared facilities, following completion of cleaning validation?
A: Analytical testing is expected at each changeover unless justified otherwise via a robust, documented Quality Risk Management (QRM) process. The QRM process should consider, at a minimum, each of the following:
- the repeatability of the cleaning process (manual cleaning is generally less repeatable than automated cleaning);
- the hazard posed by the product;
- whether visual inspection can be relied upon to determine the cleanliness of the equipment at the residue limit justified by the HBEL.
Q8. What are the requirements for conducting visual inspection as per Q&A 7?
A. When applying visual inspection to determine cleanliness of equipment, manufacturers should establish the threshold at which the product is readily visible as a residue. This should also take into account the ability to visually inspect the equipment, for example, under the lighting conditions and distances observed in the field.
Visual inspection should include all product contact surfaces where contamination may be held, including those that require dismantling of equipment to gain access for inspection and/or by use of tools (for example, mirror, light source, boroscope) to access areas not otherwise visible. Non-product contact surfaces that may retain product that could be dislodged or transferred into future batches should be included in the visual inspection.
Written instructions specifying all areas requiring visual inspection should be in place and records should clearly confirm that all inspections are completed.
Operators performing visual inspection require specific training in the process, including periodic eye sight testing. Their competency should be proven through a practical assessment.
Discussion: These two new Q&As come as quite a surprise since there was no open discussion or warning given to industry about these new topics being included in the final version of the Q&A. The opening statement in Q7 will come as a real surprise to many in the industry in that the EMA states that it now requires analytical testing after product changeover. This adds a new burden to industry, as not all manufacturers have been performing testing after every changeover once cleaning validation studies have been completed, and it undermines the validity of cleaning validation programs.
However, the alternative offered to this is to perform a documented QRM process that meets three criteria. These three criteria must include documentation of the following:
- that the cleaning process is repeatable,
- what hazard is posed by the product (presumably low),
- and the most interesting, whether visual inspection can be relied upon.
It would have been very helpful for both industry and the EMA for the agency to have solicited input on visual inspection prior to releasing a final document. Fortunately, two of this article's authors recently published an article on how to demonstrate whether visual inspection is acceptable at a facility where the cleaning process was shown to be very repeatable, the hazard level was very low, and the cleaning of the manufacturing equipment was very safe (points 1 and 2). This article also showed how to qualify a large number of personnel in a reasonable manner that is statistically valid and can be easily documented and maintained (point 3).15 Subsequent articles expanding on this work are planned.
Members of the ASTM cleaning and HBEL teams have already published a series of articles that detail how to evaluate and measure the risk in cleaning15, 17-20 and described how a cleaning control strategy could be developed that includes visual inspection.21 So, these two Q&As are very welcome.
Additionally, it should be noted that instead of performing sampling and analytical testing for residuals upon completion of validation, the cleaning team should be encouraged to set up a continued verification program using visual inspection limits outlined above, as well as trending of cleaning results. This would instill a culture of cleaning process understanding and improve knowledge management in this area [ICH Q10].
Summary
These Q&As and their updates mark an important turning point in the pharmaceutical industry regarding cleaning validation and QRM. The release of the Q&As coincides with the publication of the ASTM E3106-18 Standard Guide for Science-Based and Risk-Based Cleaning Process Development and Validation and the upcoming publication of the ASTM Standard Guide for the Derivation of Health-Based Exposure Limits (HBELs). These new standards will provide guidance to the industry on the appropriate development of HBELs, for the implementation of science- and risk-based approaches to cleaning validation, and on the use of statistical techniques for measuring the risk in cleaning and assessing the effectiveness of cleaning processes.
Recently, Graeme McKilligan, a senior GMP inspector in the Medicines and Healthcare products Regulatory Agency (MHRA), posted a statement on the MHRA blog that clarified the EMA's position on the Q&As and their relationship to cleaning validation. 22 McKilligan makes it clear that HBELs are now required and the industry should proceed with implementation:
"...it is important that those manufacturers who have not developed HBELs (such as PDE) should do so without delay. Whilst some pragmatism will be applied for manufacturers with products that present lesser hazard, those manufacturers with products likely to have lower HBEL (toward the dark orange and red area of the continuum diagram) should complete these assessments without delay (a triage approach may be helpful for prioritisation)". (emphasis added)
From the authors’ perspective this is welcome news, but, unfortunately, there has been resistance within the industry against moving to more science-based, risk-based, and statistics-based approaches to cleaning validation and setting limits, and arguments have been made for remaining with the traditional approaches. While these arguments against the science for moving to HBELs have been effectively addressed,7-12 there are other reasons for this resistance that need to be addressed as well.
Some of the resistance has come from the belief that HBELs are an unnecessary financial burden. Regardless of cost, the undeniable importance for determining HBELs is to identify any compounds that pose a high hazard to patient safety so that appropriate controls will be utilized, so this financial argument is without merit. The only way to demonstrate that a compound is not a high hazard is to derive an HBEL for it, so all medicinal products must have an HBEL. However, and as was anticipated, the initial determinations of HBELs have already shown that about 85 percent of drugs have had cleaning limits that were too low, in many cases much too low, and these have been causing unnecessary operational difficulties for many years.12 By using HBELs in an ICH Q9 risk-based approach, such as described in ASTM E3106, companies manufacturing low-hazard/low-risk compounds would be able to justify minimizing their cleaning validation efforts, reducing their operational costs and even possibly converting to using only visual inspection.21 Although there is an initial cost involved in the derivation of an HBEL, there can be significant cost benefits to be gained from implementing them.
But based on companies' experience with inspections, which are often unpredictable, most companies will still be hesitant to take advantage of such operational benefits to minimize their cleaning validation efforts or to implement visual inspection. Many companies will adopt a "wait and see" stance. There are issues with regulators and inspectors, as well. Most companies will not make any changes without knowing how inspectors are going to react to the changes. Although regulators have embraced and adopted ICH Q9, there is no clear evidence that they will accept the second principle that "the level of effort, formality and documentation of the quality risk management process should be commensurate with the level of risk." This also applies to the validation efforts (II.6 Quality Risk Management as Part of Production - Validation). As mentioned above, low-hazard/low-risk companies could reduce their validation efforts. But if inspectors don't respect this aspect of risk management in their inspections, there is little incentive for companies to adopt science-based, risk-based, and statistics-based approaches. So, regulators need to publicly support the reduction in validation efforts/documentation for lower-hazard/lower-risk companies that implement "science-, risk-, and statistics-based approaches" to cleaning. Q&As 7 and 8 are good examples of where a regulator has made such a start, and the EMA should be applauded for that.
This brings us to issues around the ongoing conflicts in regulatory guidance. The EMA has been proactive in writing the original guideline on HBELs and updating Annex 15 to require a "toxicological evaluation," which PIC/S subsequently adopted along with the guidance on HBELs. The health authorities of 49 countries are part of PIC/S, including the FDA. However, the FDA still has its original 1993 cleaning guide23 in use, which refers to the older approaches, but it has now suggested that the 2011 Process Validation Guidance applies to cleaning. 24 The FDA has also adopted ICH Q9, and this is very significant for the ASTM E3106 since it was structured on both ICH Q9 and the FDA's 2011 guidance. But it is not clear whether the FDA will follow Annex 15 or its 2011 guidance. In addition to this, there remains guidance from other regulatory organizations, such as WHO25 and Health Canada,26 that still refers to the older approaches, and many companies are confused on how to handle this. If regulators could consolidate all their guidance around the HBEL, that would be very helpful.
There has also been an argument against the need for HBELs from the standpoint of pharmacovigilance. Basically, some in industry have stated that there is no indication from the ongoing pharmacovigilance that there are any problems using the traditional limits. However, it is not possible for pharmacovigilance to reveal problems with cleaning. A cleaning failure may go completely undetected during processing, and hazardous residues may cross contaminate a product and even reach a patient.27 While the Viracept problem was detected by patients from the obvious odor, there is a strong probability that similar events have taken place and gone undetected, even though patients may have been adversely affected. How would a physician know that a complaint, or even an adverse reaction in a patient, is due to residues from some other product that the physician has no knowledge of? How would pharmacovigilance detect an adverse reaction to a cleaning validation failure? Regardless, it is poor QRM to wait for a problem to happen before taking preventive action. This is why the ASTM E3106 standard recommends that cleaning procedures are subjected to risk assessments (e.g., FMEA/FMECA) to identify possible failure modes and take preventive actions before these failures can happen. Such risk assessments should take the HBEL into consideration.15, 17
Some of the building blocks to create a triage approach for prioritization have already been put in place. The first, a toxicity scale mathematically derived directly from the HBEL, has been developed that allows HBELs to be displayed together on a scale from 0 to 10, where their relative hazards can be quickly and easily visualized.15 A process capability scale has also been developed that allows existing cleaning data to be evaluated against their HBELs and displayed together also on a scale from 0 to 10 where the relative reliability of the cleaning processes can also be quickly and easily visualized.18 These two scales provide visualization of the two main aspects of risk, that is, hazard plus exposure. Two detectability scales were also developed for total organic carbon analysis and visual inspection (but can be used for any analytical method) that can visualize the justification for these analytical methods to be used with the compound and the acceptability of any cleaning data that has been collected. Finally, these scales can be combined into a cleaning risk dashboard that can allow simple and easy visualization of the level of risk posed by any cleaning process.16 Spreadsheets have been developed to create all of these scales and display the results. They are available to the industry free of charge.28-30 Indeed, these scales should be used together, as a high hazard in itself is not necessarily a direct indicator of high risk, as the level of exposure must also be known.31 Using these spreadsheets is simple and straightforward. Implementing a triage approach using these scales could be the first step in a transitional period for companies to move from a traditional to a science-based approach to cleaning validation.
While such an approach using the HBELs would be science- and risk-based and relatively simple to implement, it should be understood that the industry rarely takes the initiative unless spurred on by regulator action. Regulators are already requesting risk assessments, and this simple approach can quickly and easily identify whether a company has cleaning risks or not.
Peer Review
The authors wish to thank our peer reviewers: Bharat Agrawal James Bergum, Ph.D.; Gabriela Cruz, Ph.D.; Mallory DeGennaro; Parth Desai; Kenneth Farrugia; Ioanna-Maria Gerostathi; Miquel Romero Obon; Laurence O'Leary; and Joel Young for reviewing this article and for their insightful comments and helpful suggestions.
References
- EMA Guideline on Setting Health Based Exposure Limits for Use in Risk Identification in the Manufacture of Different Medicinal Products in Shared Facilities, EMA/CHMP/CVMP/ SWP/169430/2012, 20 November 2014. www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2014/11/WC500177735.pdf.
- American Society for Testing and Materials (ASTM) E3106-17 "Standard Guide for Science-Based and Risk-Based Cleaning Process Development and Validation" www.astm.org.
- ASTM Work Item WK59975 - the Derivation of Health Based Exposure Limits (HBELs)
- European Medicines Agency: Questions and answers on implementation of risk-based prevention of cross-contamination in production and “Guideline on setting health-based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities" 15 December 2016 EMA/CHMP/CVMP/SWP/463311/2016
- European Medicines Agency: Questions and answers on implementation of risk-based prevention of cross-contamination in production and “Guideline on setting health-based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities" 19 April 2018 EMA/CHMP/CVMP/SWP/246844/2018
- Concept Paper on the development of toxicological guidance for use in risk identification in the manufacture of different medicinal products in shared facilities” (EMA/CHMP/SWP/598303/2011) a precursor to “Guideline on setting health-based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities” (EMA/CHMP/CVMP/SWP/169430/2012) https://www.ema.europa.eu/documents/scientific-guideline/concept-paper-development-toxicological-guidance-use-risk-identification-manufacture-different_en.pdf
- Walsh, Andrew. (2011). "Cleaning Validation for the 21st Century: Acceptance Limits for Active Pharmaceutical Ingredients (APIs): Part I", Pharmaceutical Engineering, July/August 2011. Vol. 31 (No. 4),
- Walsh, Andrew. (2011). "Cleaning Validation for the 21st Century: Acceptance Limits for Active Pharmaceutical Ingredients (APIs): Part II", Pharmaceutical Engineering, September/October 2011. Vol. 31 (No. 5)
- Walsh, Andrew, Mohammad Ovais, Thomas Altmann and Edward V. Sargent. "Cleaning Validation for the 21st Century: Acceptance Limits for Cleaning Agents", Pharmaceutical Engineering, November/December 2013. Vol. 33 (No. 6),
- Weideman et.al. (2015) "Deriving Health-Based Exposure Limits in the Pharmaceutical Industry," Contract Pharma, September 2015
- Crevoisier, M. et. al., "Cleaning Limits - Why The 10-ppm Criterion Should Be Abandoned," Pharmaceutical Technology January 2016, Vol. 40 (No. 1): 52-56
- Walsh, Andrew, Michel Crevoisier, Ester Lovsin Barle, Andreas Flueckiger, David G. Dolan, Mohammad Ovais (2016) "Cleaning Limits—Why the 10-ppm and 0.001-Dose Criteria Should be Abandoned, Part II," Pharmaceutical Technology 40 (8).
- Faria, E. C., Bercu, J. P., Dolan, D. G., Morinello, E. J., Pecquet, A. M., Seaman, C., . . . Weideman, P. A. (2016). Using default methodologies to derive an acceptable daily exposure (ADE). Journal of Regulatory Toxicology and Pharmacology, 79 Suppl 1.
- ISPE Baseline® Guide: Risk-Based Manufacture of Pharmaceutical Products: A Guide to Managing Risks Associated with Cross-Contamination. (ISPE, Tampa, FL, First Edition, 2010), Vol. 7, p. 186.
- Walsh, Andrew, Michel Crevoisier, Ester Lovsin Barle, Andreas Flueckiger, David G. Dolan, Mohammad Ovais, Osamu Shirokizawa, and Kelly Waldron "An ADE-derived Scale for Assessing the Risk of Compound Carryover in Shared Facilities," PharmaceuticalOnline May 22, 2017
- Walsh, Andrew, Thomas Altmann, Alfredo Canhoto, Ester Lovsin Barle, David G. Dolan, Andreas Flueckiger, Igor Gorsky, Jessica Graham, Ph.D., Robert Kowal, Mariann Neverovitch, Mohammad Ovais, Osamu Shirokizawa and Kelly Waldron "Measuring Risk in Cleaning: Cleaning FMEA and the Cleaning Risk Dashboard," PharmaceuticalOnline April 2018
- Best M. & Neuhauser D., "Walter A Shewhart, 1924, and the Hawthorne factory," Quality and Safety in Health Care. 2006 Apr;15(2):142-3 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2464836/.
- Walsh, Andrew, Ester Lovsin Barle, David G. Dolan, Andreas Flueckiger, Igor Gorsky, Robert Kowal, Mohammad Ovais, Osamu Shirokizawa and Kelly Waldron "A Process Capability-derived Scale for Assessing the Risk of Compound Carryover in Shared Facilities," PharmaceuticalOnline, August 2017
- Walsh, Andrew, Thomas Altmann, Alfredo Canhoto, Ester Lovsin Barle, David G. Dolan, Andreas Flueckiger, Igor Gorsky, Robert Kowal, Mariann Neverovitch, Mohammad Ovais, Osamu Shirokizawa and Kelly Waldron "An MSSR-derived Scale for Assessing the Detectability of Visual Inspection," PharmaceuticalOnline, December 2017
- Walsh, Andrew, Thomas Altmann, Alfredo Canhoto, Ester Lovsin Barle, David G. Dolan, Andreas Flueckiger, M.D., Igor Gorsky, Robert Kowal, Mariann Neverovitch, Mohammad Ovais, Osamu Shirokizawa and Kelly Waldron. "A Swab Limit-Derived Scale For Assessing The Detectability Of Total Organic Carbon Analysis," PharmaceuticalOnline, January 2018
- Andrew Walsh, Dongni (Nina) Liu and Mohammad Ovais, "Justification and Qualification of Visual Inspection for use in Cleaning Validation for a Low Risk, Multi-Product Facility," PharmaceuticalOnline, August 2018
- https://mhrainspectorate.blog.gov.uk/2018/10/22/cross-contamination-control-and-health-based-exposure-limits-hbel-qas/
- Food and Drug Administration Guide to Inspections: Validation of Cleaning Processes
- International Pharmaceutical Quality October 29, 2015 (Clarifying Questions Upfront is Key in Process Validation, US and EU PV Principles in Alignment —CDER’s McNally) https://www.ipqpubs.com/2015/10/29/clarifying-questions-upfront-is-key-in-process-validation-us-and-eu-pv-principles-in-alignment-cders-mcnally-stresses/
- World Health Organization - WHO Technical Report Series, No. 937, 2006 Annex 4 Supplementary guidelines on good manufacturing practices: validation, Appendix 3 Cleaning validation
- Health Canada: Health Products and Food Branch Inspectorate Guidance Document - Cleaning Validation Guidelines GUIDE-0028
- Killilea, M.C., “Cleaning Validation: Viracept, 2007,” Journal of Validation Technology, November 2012, Vol. 18, Issue 4
- Spreadsheet to Create a Toxicity Scale from HBELs - https://www.researchgate.net/publication/324259996_Spreadsheet_to_Create_a_Toxicity_Scale_from_HBELs
- Spreadsheet to Create a Process Capability Scale from Cpu Data - https://www.researchgate.net/publication/324260079_Spreadsheet_to_Create_a_Process_Capability_Scale_from_Cpu_Data
- Spreadsheet for create Detectability Scales from TOC and Visual Inspection Detection Limits - https://www.researchgate.net/publication/324260010_Spreadsheet_for_create_Detectability_Scales_from_TOC_and_Visual_Inspection_Detection_Limits
- Lovsin-Barle, Ester and Andrew Walsh, "Are high potency active pharmaceutical ingredients (HPAPI) also high risks for cross-contamination?" December 2017Chimica oggi 35(6)