
Developing A Science-, Risk-, & Statistics-Based Approach To Cleaning Process Development & Validation
By Thomas Altmann, Alfredo Canhoto, Michel Crevoisier, Igor Gorsky, Robert Kowal, Mariann Neverovitch, Mohammad Ovais, Osamu Shirokizawa, and Andrew Walsh
 Part of the Cleaning Validation For The 21st Century series1
Part of the Cleaning Validation For The 21st Century series1
Cleaning manufacturing equipment to prevent cross contamination of pharmaceutical products is a fundamental aspect of GMPs. Validation of cleaning processes has been required within cGMP industries for a long time and is recognized as an important activity to establish that product cross contamination is controlled to ensure patient safety and product quality.
While cleaning, in and of itself, is a relatively simple process, the pressures of inspection scrutiny and the reactionary programs created by industry to address regulatory concerns have transformed the validation of cleaning into a complex, expensive, and time-consuming activity. From a simple project management analysis, the time that would be required to perform cleaning validation for a facility with multiple products, multiple pieces of equipment, and multiple cleaning procedures can easily run into years.
Figure 1 shows a very aggressive, and a clearly hard to accomplish, timeline for concurrently performing three runs for only three cleaning validations allowing only one day between runs and ignoring analysis time and other items. Despite this, the timeline is still six months.
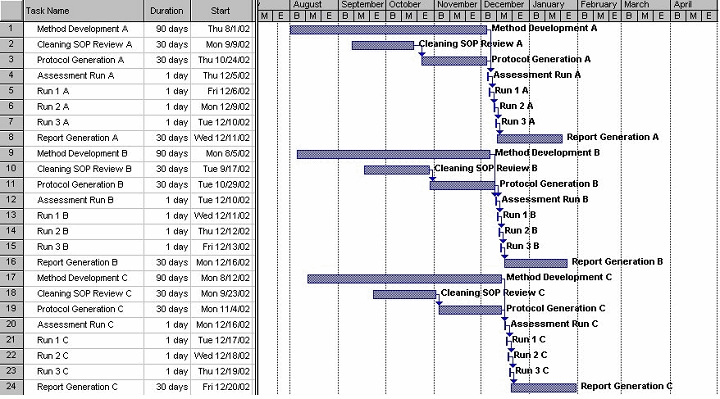
Figure 1: Hypothetical timeline for cleaning validation of three products
Considering that cleaning validation runs cannot be scheduled and performed every day and the need for method development, protocol development, laboratory analysis, and report writing, it is clear that cleaning validation consumes a considerable amount of resources.
Consequently, companies have made various efforts to reduce cleaning activities, such as dedicating equipment or converting to disposable items, but these strategies have their own inefficiencies and costs. Companies have also resorted to strategies such as product grouping, equipment grouping, matrixing, and bracketing to reduce the amount of cleaning they validate, sometimes without acceptable justification. Many companies today validate the cleaning of only one or two "hardest-to-clean" products, selecting them based on the solubility of the API or because the calculated limit is lowest, even though these may not be truly justifiable criteria. Even with such efforts, part of the reality has been that, for all intents and purposes, cleaning validation never seems to be completed. The EU guideline “Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use,” Annex 15 outlines in Section 10 that for a worst case product approach a scientific rationale should be provided.2 It also outlines criteria for determining the worst case, and these criteria may include solubility, cleanability, toxicity, and potency. This approach makes determining a worst case situation even more complex.
As with many things, the pharmaceutical industry has tended to understand cleaning almost entirely in its relation to regulatory expectations. In particular, cleaning has become closely associated with “process validation.” In the late 80s/early 90s, the FDA, as well as other regulatory agencies, began to view cleaning as a process that needed to be validated.3 At the same time, several legal decisions concerning cleaning made during the resolution of the well-known Barr Labs case solidified this viewpoint.4 Consequently, companies set about validating existing cleaning procedures without questioning whether the procedures were the most effective or optimal, or even if they were using an appropriate cleaning agent. The cleaning procedures that were subsequently validated may not have been the best choice for their situations.
Cleaning validation incorporates the traditional preapproved protocol, with predetermined acceptance criteria and a three-run process validation approach. Because of the traditional approach, the industry also struggled over how to set the required predetermined acceptance criteria. This process validation approach was adopted without ever asking if three cleaning validation runs and predetermined acceptance criteria were appropriate for the validation of cleaning. Based on that reason, Annex 15 also outlines in Section 10 that the cleaning procedure should be evaluated an appropriate number of times (based on a risk assessment) and meet the acceptance criteria in order to prove that the cleaning procedure is validated.
All these issues underscore the need for effective and efficient cleaning programs that focus efforts and resources where they provide the most value.
Since 2001, there have been many new, and for this highly conservative industry, radical movements from both regulators and within the industry itself. Examples coming from the FDA include “GMPs for the 21st Century,”5 quality by design (QbD),6 process analytical technology (PAT),7 and the agency’s 2011 guideline on process validation.8 Globally, the new International Conference on Harmonisation's guidelines, in particular Q8 and Q9,9 are major forces driving change in the industry. Movements within pharmaceutical manufacturing itself include lean manufacturing, Six Sigma, and operational excellence (OpEx), which have grown out of the pressures to reduce costs and to better supply the market. These “planets” have aligned to create a tide drawing the industry toward science-based, risk-based, statistics-based, and cost-effective approaches to ensuring patient safety and product quality during pharmaceutical development and manufacturing. As one of the critical processes in manufacturing, cleaning and its validation can benefit from all these initiatives.
The introduction of the acceptable daily exposure (ADE) in 2010 provided a tool that could be used for setting science-based acceptance criteria for the cleanliness of equipment.10 Several subsequent publications have revealed how replacing the traditional approaches to setting acceptance criteria with an approach based on the ADE leads to better patient safety and can reduce the validation effort for lower-risk situations,11-14 and a recent publication discussed how an ADE-derived scale can be used to easily and visually evaluate the risks of cross contamination in manufacturing facilities, including for cleaning.15
Cleaning validation programs and master plans could benefit from a risk-based approach to their design and management. Cleaning procedures could benefit through a statistics-based QbD approach resulting in safer and more reliable procedures, and the analytical methods used in cleaning could benefit from PAT, resulting in faster turnaround of equipment. Many of the techniques used in lean manufacturing, Six Sigma, and operational excellence could be used to reduce the time and effort spent, improve the results obtained during cleaning validations, and provide statistics-based means for evaluating and controlling cleaning processes. Perhaps even cleaning, which is certainly a process, should be looked at and evaluated in the manner being suggested in the FDA’s 2011 process validation guidance; indeed, the FDA believes that this guidance is applicable to cleaning.16
The authors believe focusing industry efforts where the risks are high will increase patient safety and reducing efforts where the risks are low will ease the regulatory burden on industry and improve operational efficiencies overall.
Regulations And Current Guidance And Their Application To Cleaning
This section explores in more detail how the regulations and current guidance mentioned above provide direction on how to implement these approaches to cleaning.
The requirements in 21 CFR 211.67(a) state that “Equipment and utensils shall be cleaned, maintained, and sanitized at appropriate intervals to prevent malfunctions or contamination that would alter the safety, identity, strength, quality, or purity of the drug product beyond the official or other established requirements.”17
Similarly, 21 CFR 111.27(d) states “You must maintain, clean, and sanitize, as necessary, all equipment, utensils, and any other contact surfaces used to manufacture, package, label, or hold components or dietary supplements.”18
21 CFR 820.70(e) also states “Contamination control. Each manufacturer shall establish and maintain procedures to prevent contamination of equipment or product by substances that could reasonably be expected to have an adverse effect on product quality.”19
From these statements, several required elements of a cleaning program can be determined: the scope of cleaning, a required schedule for maintenance, and targets to achieve. In order to alter the “identity,” “strength,” or “purity” of a product, certainly gross contamination would be required. Such high levels should not be found after cleaning. However, in some cases, process residues below the order of gross contamination may still affect patient safety and possibly product quality. One goal of a cleaning program is to verify that no gross contamination remains after cleaning and that any residues that do remain do not jeopardize the safety of the patient or quality of the next product.
Now let's look at some of the many regulatory guidances that have come out since 2001. While some of them have some degree of application to cleaning, the two guidances that have the most applicability to cleaning are ICH Q9 and the FDA's 2011 process validation guidance.
ICH Q9 Guidance
ICH Q9 outlines basic principles and examples of tools for quality risk management that can be applied to pharmaceutical processes. In ICH Q9 we find two primary principles of quality risk management:9
- The evaluation of the risk to quality should be based on scientific knowledge and ultimately link to the protection of the patient; and
- The level of effort, formality, and documentation of the quality risk management process should be commensurate with the level of risk.
If we apply these principles to cleaning, it is apparent that the risks the cleaning processes may present to patient safety and product quality should be scientifically assessed. The extent of any activities, such as cleaning development, cleaning verification, cleaning validation, monitoring, etc., should then be driven by the level of risk presented. The implementation of these principles offers serious potential for developing useful, effective, and efficient cleaning programs.
In fact, Annex II, “Potential Applications for Quality Risk Management,” subsection 6, "Quality Risk Management as Part of Production" under Validation states, "To identify the scope and extent of verification, qualification and validation activities (e.g., analytical methods, processes, equipment and cleaning methods," which clearly encourages the use of ICH Q9 for developing a cleaning validation program. Annex II subsection II.4 "Quality Risk Management for Facilities, Equipment and Utilities" also states that ICH Q9 principles can be applied to setting "acceptable (specified) cleaning validation limits." A precedent for implementing ICH Q9, as it pertains to cleaning, has already been set for this in the International Society for Pharmaceutical Engineering's (ISPE) Risk-Based Manufacturing of Pharmaceutical Products (Risk-MaPP) Baseline® Guide.10 Figure 2 shows an overview of the ICH Q9 quality risk management process.

Figure 2: Overview of a typical quality risk management process
As ICH Q9 suggests, the risk management process can also be applied to the cleaning of all manufacturing equipment. Consequently, the risk assessment process should be used to derive criteria that can assist in decision making and control the risks to the patient. For a cleaning process, this should be a systematic and documented process to:
- identify the hazard (e.g., cleaning process residues)
- assess the severity of the cleaning process residues
- evaluate means to detect the cleaning process residues
- determine the levels of cleaning process residues
- support the implementation and maintenance of appropriate controls.
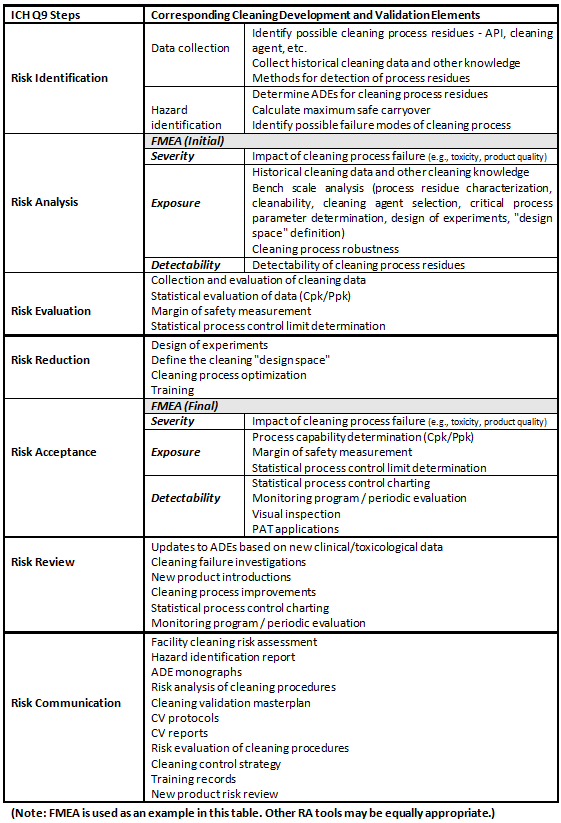
Risk controls should be commensurate with the level of risk. The ultimate decision on the appropriate controls may rely on both qualitative and quantitative data. The risk assessment should be documented and should include a discussion of all inherent assumptions and limitations. Table 1 shows how cleaning process development and validation maps to the ICH Q9 process.
Table 1: Map of ICH Q9 Elements to Cleaning
Process Validation: General Principles And Practices
The FDA’s process validation guidance8 aligns with the product life cycle concept and with existing FDA guidance on ICH Q8-Q10 and also describes concepts that are directly applicable to cleaning and cleaning validation. We can simply add "cleaning" to the elements of the process validation guidance as shown below.
- Cleaning Process Design — Building and capturing process knowledge and understanding
- Application of design of experiment to cleaning
- Multifactorial interactions
- Using risk analysis tools to screen potential variables
- Cleaning Process Qualification
- Use of statistical methods in analyzing all collected cleaning data
- Continued Cleaning Process Verification
- Use of statistical process control techniques
- Continuous Improvement
- Use of historical data (monitoring, etc.) or technological advances for improvement of cleaning processes
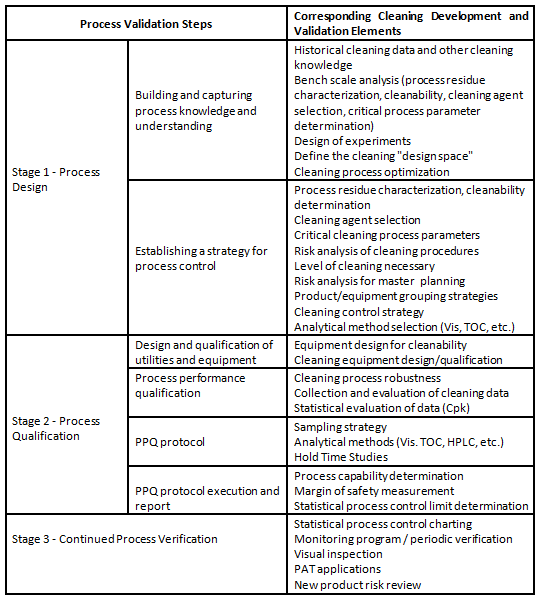
The elements of the process validation guideline can be easily worked into a framework for a science-, risk-, and statistics-based approach to cleaning. Table 2 shows how cleaning process development and validation maps to the FDA's process validation guidance.
Table 2: Map of FDA's Process Validation Guidance Elements to Cleaning
cGMPs For The 21st Century Guidance
In the FDA guidance “Pharmaceutical cGMPs for the 21st Century — A Risk-Based Approach”5 we see four principles that have particular relevance to cleaning:
- Encourage the early adoption of new technological advances by the pharmaceutical industry.
- Facilitate industry application of modern quality management techniques, including implementation of quality systems approaches, to all aspects of pharmaceutical production and quality assurance.
- Encourage implementation of risk-based approaches that focus both industry and agency attention on critical areas.
- Ensure that regulatory review, compliance, and inspection policies are based on state-of the-art pharmaceutical science.
Applying these principles to cleaning, it follows that the extent of any activities, such as cleaning development and cleaning validation, should be driven by the level of risk presented and that the use of modern technology is encouraged.
PAT Guidance
The FDA guidance “PAT - A Framework for Innovative Pharmaceutical Development, Manufacturing, and Quality Assurance”7 states (again adding cleaning):
- A desired goal of the PAT framework is to design and develop well-understood cleaning processes that will consistently ensure a predefined quality at the end of the cleaning process. Such cleaning procedures would be consistent with the basic tenet of quality by design and could reduce risks to quality and regulatory concerns while improving efficiency.
- Reducing cleaning cycle times by using on-, in-, and/or at-line measurements and controls
In the PAT guidance we find that, as a process, cleaning should be designed, developed, and well understood, and the use of on-, in-, and at-line measurements and controls is encouraged.
Quality By Design
Although the quality by design initiative as described in ICH Q8-Annex 1 addresses product manufacturing processes, there are principles there that can be applied to cleaning processes as well, such as (once again adding cleaning):6
- Selecting an appropriate cleaning process.
- Identifying a cleaning control strategy (CS).
- A systematic evaluation, understanding and refining of the cleaning process, including:
- Identifying, through e.g., prior knowledge, experimentation, and risk assessment, the material attributes and cleaning process parameters that can have an effect on cleaning critical quality attributes (CQAs);
- Determining the functional relationships that link material attributes and cleaning process parameters to cleaning CQAs.
- Using the enhanced cleaning understanding in combination with quality risk management to establish an appropriate control strategy which can, for example, include a proposal for design space(s) and/or real-time release.
Using a systematic approach such as those described in the Q8-Annex 1 could enable continual improvement and innovation of cleaning processes without being locked into previously validated parameters and restricted by onerous change control procedures.
ICH Q7 Guidance
ICH Q7 Section 5.2.5 states that “Acceptance criteria for residues and the choice of cleaning procedures and cleaning agents should be defined and justified".20
Through the use of the word “justified,” this simple sentence implies that science-based approaches should be employed in setting acceptance criteria for cleaning development and cleaning validation. The risk-based approach to cleaning validation is further recommended in point 12.70:
"In general, cleaning validation should be directed to situations or process steps where contamination or carryover of materials poses the greatest risk to API quality."
Although ICH Q7 applies specifically to APIs, the concept that science-based and risk-based approaches should be employed in cleaning development and cleaning validation can be extended to all pharmaceuticals.
Annex 15 Of The EU GMP Guide
Annex 15 states that:2
“Limits for the carryover of product residues should be based on a toxicological evaluation to determine the product specific Permitted Daily Exposure (PDE) value1. The justification for the selected PDE value should be documented in a risk assessment which includes all the supporting references”
1 See EMA Guideline on setting health based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities
As in ICH Q7, the use of the word “justification” implies that science-based and risk-based approaches should be employed in setting acceptance criteria for cleaning development and cleaning validation. Note: Annex 15 is applicable to pharmaceutical products.
Operational Excellence And Six Sigma
Operational excellence can be defined as conducting business in a manner that satisfies customer demand, improves quality, and generates higher yields, faster throughput, and less waste. Six Sigma can be defined as a disciplined, data-driven approach and methodology for eliminating defects in any process.
These two approaches provide statistical tools to improve processes and increase quality. Since cleaning is a process that can be measured, these techniques can be effectively used to improve the cleaning process and enhance the safety and quality of pharmaceutical products.
Summary
The guidance discussed above can be applied to create a new approach to cleaning and cleaning validation that is based on science, risk, and statistics. It offers clear ways of making sensible changes in cleaning that would reduce the complexity, lower the costs, and shorten the process while providing an even higher probability that cleaning of pharmaceutical manufacturing equipment has been effective. By implementing a truly science-based approach, such as the use of the ADE for risk analysis, with appropriate risk assessments, and with cleaning process development in place, a streamlined cleaning program may be readily developed that ensures patient safety and product quality while lightening the regulatory burden on industry.
Peer Reviewers: James Bergum, Ph.D.; Pernille L. Damkjær; Mallory DeGennaro; Michael Hrytsack, Ph.D.; Michael Schousboe; and Joel Young
References:
- This article is an excerpt and adaption from "Science, Risk & Statistics-based Guide to Cleaning Process Development and Validation" (United States Copyright Office Number TXu001950765) that was submitted in August 2013 to the International Society for Pharmaceutical Engineering (ISPE) for potential publication as a Baseline Guide. Portions of this article were previously published in Pharmaceutical Engineering November/December 2011 Vol. 31 No. 6.
- EudraLex - Volume 4 - Good Manufacturing Practice (GMP) guidelines - Annex 15 Qualification and validation.
- FDA Guide to Inspections Validation of Cleaning Processes, Section IV. Evaluation of Cleaning Validation, July 1993, U.S. Food and Drug Administration (FDA), www.fda.gov.
- United States vs. Barr Laboratories, Inc. Civil Action No. 92-1744, U.S. District Court for the District of New Jersey: 812 F. Supp. 458. 1993 US Dist. Lexis 1932; 4 February 1993, as amended 30 March 1993.
- Pharmaceutical CGMPs for the 21st Century Industry – A Risk-Based Approach: Final Report, September 2004, U.S. Food and Drug Administration (FDA), www.fda.gov.
- International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use, ICH Harmonised Tripartite Guideline, Pharmaceutical Development – Q8(R2), August 2009, www.ich.org.
- FDA Guidance for Industry: PAT – A Framework for Innovative Pharmaceutical Development, Manufacturing, and Quality Assurance, September 2004, U.S. Food and Drug Administration (FDA), www.fda.gov.
- FDA Guidance for Industry: Process Validation - General Principles and Practices January 2011, U.S. Food and Drug Administration (FDA), www.fda.gov.
- International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use, ICH Harmonised Tripartite Guideline, Quality Risk Management – Q9, Step 4, 9 November 2005, www.ich.org.
- ISPE Baseline® Guide: Risk-Based Manufacture of Pharmaceutical Products (Risk-MaPP), International Society for Pharmaceutical Engineering (ISPE), First Edition, September 2010.
- Walsh, A., “Cleaning Validation for the 21st Century: Acceptance Limits for Active Pharmaceutical Ingredients (APIs): Part I,” Pharmaceutical Engineering, July/August 2011, Vol. 31, No. 4, pp. 74- 83.
- Walsh, A., “Cleaning Validation for the 21st Century: Acceptance Limits for Active Pharmaceutical Ingredients (APIs): Part II,” Pharmaceutical Engineering, September/October 2011, Vol. 31, No. 5, pp 44-49.
- Walsh, A., Ovais, M., Altmann, T., and Sargent, E.V., “Cleaning Validation for the 21st Century: Acceptance Limits for Cleaning Agents,” Pharmaceutical Engineering, November/December 2013, Vol. 33, No. 6, pp 12-24.
- Walsh, Andrew, Michel Crevoisier, Ester Lovsin Barle, Andreas Flueckiger, David G. Dolan, Mohammad Ovais, "Cleaning Limits—Why the 10-ppm and 0.001-Dose Criteria Should be Abandoned, Part II," Pharmaceutical Technology 40 (8) 2016.
- Walsh, Andrew, Ester Lovsin Barle, Michel Crevoisier, David G. Dolan, Andreas Flueckiger, Mohammad Ovais, Osamu Shirokizawa, and Kelly Waldron, "An ADE-Derived Scale For Assessing Product Cross-Contamination Risk In Shared Facilities," Pharmaceuticalonline.com, May 2017.
- "Clarifying Questions Upfront is Key in Process Validation, US and EU PV Principles in Alignment —CDER’s McNally" in http://www.ipqpubs.com/uncategorized/clarifying-questions-upfront-is-key-in-process-validation-us-and-eu-pv-principles-in-alignment-cders-mcnally-stresses/.
- 21 CFR Part 211.67 – Current Good Manufacturing Practice for Finished Pharmaceuticals, Equipment Cleaning and Maintenance, U.S. Code of Federal Regulations, U.S. Food and Drug Administration (FDA), www.fda.gov.
- 21 CFR Part 111.27 – Current Good Manufacturing Practice in Manufacturing, Packaging, Labeling, or Holding Operations for Dietary Supplements, Equipment and Utensils; What Requirements Apply to the Equipment and Utensils that You Use? U.S. Code of Federal Regulations, U.S. Food and Drug Administration (FDA), www.fda.gov.
- 21 CFR Part 820.70 – Quality System Regulation, Production and Process Controls, U.S. Code of Federal Regulations, U.S. Food and Drug Administration (FDA), www.fda.gov.
- International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use, ICH Harmonised Tripartite Guideline, Quality Risk Management – Q9, Step 4, 9 November 2005, www.ich.org.
About The Authors:
Thomas Altmann is RD&E global technical manager – life sciences at Ecolab. He has 18 years' experience in consulting in the pharmaceutical and biotechnology industry, as well as API production related to cleaning, validation, and disinfecting of product contact surfaces.
Alfredo Canhoto, Ph.D., is a senior manager of quality assurance validation for Alnylam Pharmaceuticals and has expertise in cleaning development and validation.
Michel Crevoisier retired from Novartis Pharma in 2015. In his last position, he was a senior QA expert for qualification and validation.
Igor Gorsky is a senior consultant at ConcordiaValsource and a frequent speaker/writer on topics such as cleaning validation, critical utilities, process scale-up and validation, and knowledge management.
Robert Kowal is a retired cleaning validation subject matter expert (SME) for Johnson & Johnson.
Mariann Neverovitch is a research scientist and cleaning validation SME in Bristol-Myers Squibb's analytical strategic operations.
Mohammad Ovais is scientific affairs manager for Xepa-Soul Pattinson (M) Sdn Bhd, Malaysia and a long-time student of cleaning validation.
Osamu Shirokizawa is a director and senior consultant of Life Scientia Limited, a pharmaceutical engineering and consultancy firm.
Andrew Walsh is president of the Center for Pharmaceutical Cleaning Innovation, a non-profit research and educational organization.