
Demonstrating Value And Securing Patient Access For Cell And Gene Therapies
By Mathias Cousin, Sanjay Srivastava, Amit Agarwal, and Rebecca Brian, Deloitte

Part 1 in a three-part series
Because the parameters for ex vivo cell and gene therapy manufacturing differ considerably from chemical drug production, life sciences companies are likely to encounter challenges along key stages of the product life cycle.
Ex vivo cell and gene therapies were among the first cell and gene therapies to gain FDA approval and could extend patients’ lives significantly. Successful applications have been seen in oncology, where therapies can counteract how tumors suppress the immune system to effectively attack tumor cells.1 Chimeric antigen receptors added to T cells (CAR-T) and injected back into the patient are demonstrating encouraging results: Early studies show CD-19-targeted CAR-T therapy treatments from Novartis, Juno, and Kite provide positive results in a range of liquid cancers. Patients from some of these trials have experienced greater than one-year remissions with persistence of CAR T cells.2
CAR-T, a cell therapy technology, represents the first approved and currently largest share of the global cell and gene therapy market. The United States and China are the leaders in the development of cell therapies; the United States alone has the most therapies under investigation (300+). The FDA expects 200 cell and gene therapy investigational new drug applications each year by 2020, with 15 to 20 approvals each year by 2025.3
Two CAR-T products are currently on the market, and global sales of approved ex vivo cell and gene therapies (including CAR-T) are expected to reach $3 billion in 2022 (Figure 1).
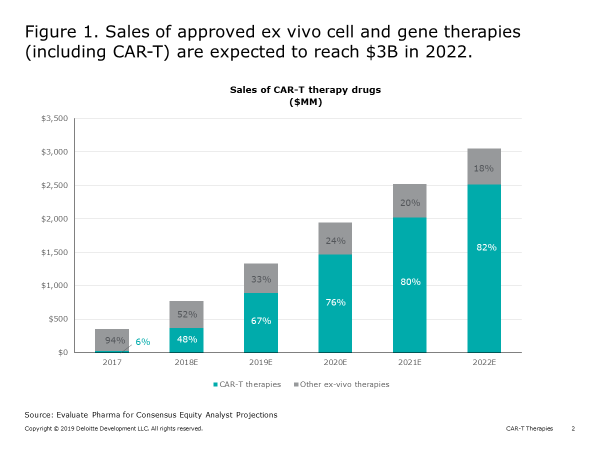
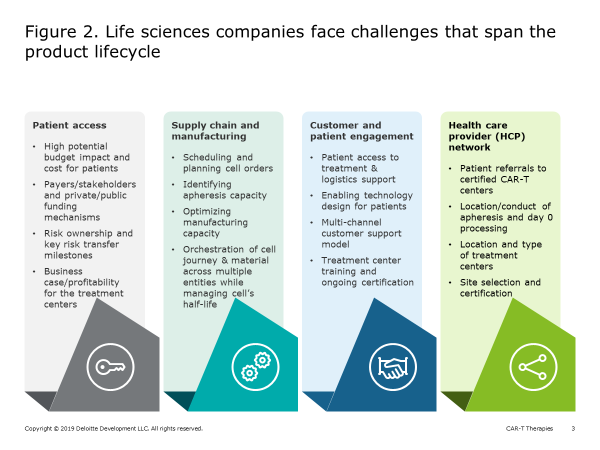
While CAR-T holds great promise for transforming the treatment for cancer, and other autologous therapies (leveraging similar vectors) may do the same with other diseases (e.g., sickle cell anemia), achieving commercial success for an individual cell and gene therapy has proven difficult. Because the parameters for ex vivo gene and cell therapy manufacturing differ considerably from chemical drug production, life sciences companies are likely to encounter potential challenges in four key areas (Figure 2):
- Patient access
- Supply chain and manufacturing
- Customer and patient engagement
- Healthcare provider (HCP) network
This three-part article series explores major challenges and lessons learned for biopharma companies developing and launching new cell and gene therapies. We begin our review with a particularly thorny issue: overcoming “sticker shock” and securing patient access despite the limitations of current payment models.
Patient Access Challenges
A major hurdle for cell and gene therapy manufacturers is securing patient access and establishing the appropriate funding/reimbursement mechanisms so providers are willing to prescribe these highly customized, clinically intensive, and costly drugs. Potential barriers to access include sticker shock for patients and payers; uncertainty about the long-term value of the drug (e.g., the duration of its effect); lack of analogs on financial flows between health systems, manufacturers, and payers; lack of understanding of proper coding guidance for health systems; and stringent payer authorization requirements on reimbursement.
Demonstrating a product’s value to the healthcare ecosystem and, specifically, to patients is at the heart of manufacturers’ efforts to secure patient access. This focus includes incorporating value evidence requirements into clinical trials design, demonstrating the product’s significant improvement in short-term clinical outcomes over the standard of care and demonstrating long-term durable efficacy, and anchoring product pricing and reimbursement to a demonstrated value proposition to patients. In an example of the anchoring of product pricing and reimbursement, Boston-area Bluebird Bio is developing plans to sell its first gene-replacement therapy, for a rare inherited blood disease, on a five-year installment plan. Each annual payment will be contingent on the treatment’s continued effectiveness. Further, the product’s price will not be higher than the company’s calculated value to patients, which leaves the healthcare system with a significant cost saving.4
Cell and gene therapies are expensive and potentially one-off treatments. Because the majority are breakthrough therapies, they may lack longitudinal clinical outcomes data on the duration of their effect due to shortened clinical trials. In some cases, the therapy may have a high budget impact on the healthcare system. To address these challenges, manufacturers have developed novel tactics to engage with key public payers, private payers, and medical associations. Manufacturers have made direct contacts with government agencies (e.g., CMS) and have worked closely with payers and policymakers to provide innovative risk-sharing arrangements. CMS recently proposed that Medicare cover FDA-approved CAR-T therapies when offered in a CMS-approved registry or clinical study, as long as data is collected from the patients for at least two years after treatment. CMS plans to use the data to decide which treatments provide the most patient benefits and which CAR-T therapies to fund in the future.5
While ex vivo cell and gene therapy reimbursement mechanisms are still being defined, manufacturers have sought to work with healthcare system stakeholders to solve for access issues, for example, by developing and prioritizing payer engagement plans based on key access variables such as a therapy’s perceived clinical and economic value, competitive position, and payer scrutiny of a disease area. They have also worked to define the role of the health system in reimbursement flows (i.e., payer to health system to manufacturer, payer directly to manufacturer) and to better understand key incentives for those players that would drive adoption. And because cell and gene therapy, as compared to traditional pharma, requires significantly greater interaction with hospitals/treatment centers over the course of treatment delivery, manufacturers have acknowledged that it is critical to work with these providers throughout the patient journey.
Patient Access: Lessons Learned
Based on our observations of early cell and gene therapy commercialization efforts, biopharma companies could incorporate the following into their patient access strategies, with the goal of improving new product uptake and commercial performance:
- Define and implement a robust informatics capability to collate and correlate large data sets obtained from disparate sources — electronic medical record, manufacturing, and clinical outcomes data to help identify the combination of factors that lead to successful treatment.
- Integrate patient access considerations into the therapy’s clinical development plan/target product profile.
- Engage early and often with regulatory entities to shape the treatment policy environment so the therapy’s value is broadly recognized.
- Focus the therapy’s value narrative to payers on how it alleviates the healthcare system’s clinical and cost burdens and provides value to patients.
- Generate cost-effectiveness data ahead of a therapy’s launch to sustain its value narrative.
- Quantify the value generated by the product to different healthcare stakeholders and be prepared to offer pricing that is consistent with that value. Consider offering value-based contracting (e.g., performance guarantees, installment payments) and assume risk on endpoints where payer/providers lack confidence (e.g., duration of effect).
Subsequent articles in our series will explore cell and gene therapy challenges and lessons learned in the life cycle areas of supply chain and manufacturing and HCP networks.
References:
- Top 10 health care innovations: Achieving more for less, Deloitte Center for Health Solutions, 2016, https://www2.deloitte.com/content/dam/Deloitte/global/Documents/Life-Sciences-Health-Care/gx-lshc-top-10-health-care-innovations-web-friendly.pdf
- Colin White, Robert Jeng, Jolene Lau, “Immuno-Oncology Overview,” DataMonitor Healthcare, June 12, 2015, https://www.futureofoncology.com/wp-content/uploads/2015/06/Datamonitor-Healthcare-Immuno-Oncology-Product-Brochure.pdf, accessed July 19, 2016
- Deloitte analysis of data from clinicaltrials.gov database
- “Biotech Proposes Paying for Pricey Drugs by Installment,” The Wall Street Journal, January 8, 2019, https://www.wsj.com/articles/biotech-proposes-paying-for-pricey-drugs-by-installment-11546952520
- “Medicare would cover CAR-T therapies under new CMS proposal, MarketWatch, February 15, 2019, https://www.marketwatch.com/story/medicare-would-cover-car-t-cell-therapies-under-new-cms-proposal-2019-02-15
About The Authors:
 Mathias Cousin is a senior manager in the Life Sciences strategy practice of Monitor Deloitte, and one of the leaders of Deloitte’s biotech, and next generation therapy practices. He is passionate about how technological changes will impact the life sciences industry and potentially disrupt existing players. During his consulting tenure, Mathias has led engagements in corporate, BU, and commercial strategy, both in pre- and post-launch environments for life sciences companies. He has worked across industries and geographies.
Mathias Cousin is a senior manager in the Life Sciences strategy practice of Monitor Deloitte, and one of the leaders of Deloitte’s biotech, and next generation therapy practices. He is passionate about how technological changes will impact the life sciences industry and potentially disrupt existing players. During his consulting tenure, Mathias has led engagements in corporate, BU, and commercial strategy, both in pre- and post-launch environments for life sciences companies. He has worked across industries and geographies.

 Amit Agarwal is a managing director in Deloitte Consulting’s life sciences practice. He has more than 25 years of management consulting experience and has led multiple projects in both strategy and operations. Agarwal co-leads the Next Gen Therapy practice, which focuses on advising clients on taking advantage of and/or developing strategic responses to new disruptive technologies which leapfrog older business models and establish new clinical practices. Agarwal has worked with life sciences clients in the U.S., Europe, and Asia on high-impact projects. He holds an MBA in finance and technological innovation from the MIT Sloan School of Management and a bachelor’s degree in history and pre-med from Occidental College.
Amit Agarwal is a managing director in Deloitte Consulting’s life sciences practice. He has more than 25 years of management consulting experience and has led multiple projects in both strategy and operations. Agarwal co-leads the Next Gen Therapy practice, which focuses on advising clients on taking advantage of and/or developing strategic responses to new disruptive technologies which leapfrog older business models and establish new clinical practices. Agarwal has worked with life sciences clients in the U.S., Europe, and Asia on high-impact projects. He holds an MBA in finance and technological innovation from the MIT Sloan School of Management and a bachelor’s degree in history and pre-med from Occidental College.
 Rebecca Brian is a managing director in Monitor Deloitte’s life sciences practice, with more than 15 years of experience advising clients in the biotech/pharma sector. Her focus is building commercial strategies anchored in deep customer insight, and helping organizations translate these into action and outcomes. Brian leads engagements that address a range of strategic issues, including new product commercialization, patient and customer experience, driving growth for in-line products, and building commercial capabilities. Brian has experience across a wide range of therapeutic areas, with a particular focus on oncology and rare disease. She holds an MBA from the Stanford University Graduate School of Business.
Rebecca Brian is a managing director in Monitor Deloitte’s life sciences practice, with more than 15 years of experience advising clients in the biotech/pharma sector. Her focus is building commercial strategies anchored in deep customer insight, and helping organizations translate these into action and outcomes. Brian leads engagements that address a range of strategic issues, including new product commercialization, patient and customer experience, driving growth for in-line products, and building commercial capabilities. Brian has experience across a wide range of therapeutic areas, with a particular focus on oncology and rare disease. She holds an MBA from the Stanford University Graduate School of Business.
As used in this document, “Deloitte” means Deloitte Consulting LLP, a subsidiary of Deloitte LLP. Please see www.deloitte.com/us/about for a detailed description of our legal structure. Certain services may not be available to attest clients under the rules and regulations of public accounting.
This publication contains general information only and Deloitte is not, by means of this publication, rendering accounting, business, financial, investment, legal, tax, or other professional advice or services. This publication is not a substitute for such professional advice or services, nor should it be used as a basis for any decision or action that may affect your business. Before making any decision or taking any action that may affect your business, you should consult a qualified professional advisor. Deloitte shall not be responsible for any loss sustained by any person who relies on this publication.