
Best Practices For Compliance With The New Elemental Impurities Requirements
By Gregory P. Martin, Complectors Consulting LLC

New requirements for control of elemental impurities went into effect at the beginning of 2018, replacing the longstanding requirements for heavy metals. Elemental impurities guidances — International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) Q3D, U.S. Pharmacopeia (USP) General Chapter <232>, European Pharmacopeia (EP) 5.20 — are based on modern safety considerations specific to the elements identified. Limits are significantly lower for the four most toxic metals than the previous limits for heavy metals. Fortunately, the requirements in the various guidances are essentially harmonized. By contrast, the older heavy metals requirements used a procedure that detected a group of metals but was not specific for each one, and the limits were established arbitrarily and were inconsistent from material to material. Due to the lower limits, the new tests for elemental impurities use more expensive technology (typically inductively coupled plasma – optical emission spectroscopy [ICP-OES] or inductively coupled plasma – mass spectrometry [ICP-MS]) than the heavy metals test, which uses essentially the same technology (wet chemistry with visual endpoint) that was introduced over 100 years ago. The new guidances address not only the most toxic metals, but also a number of elements that may be introduced intentionally, bringing the total number of elements that are addressed to 24. The requirements are shown in Table 1.
Table 1: Permitted Daily Exposures for Elemental Impurities
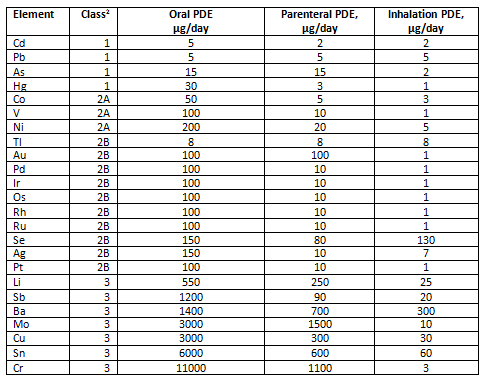
Adapted from International Conference on Harmonisation GUIDELINE FOR ELEMENTAL IMPURITIES Q3D Current Step 4 version (December 16, 2014)
Options For Control
Elemental impurities must be controlled in the drug product, but that does not necessarily mean routine testing of each lot of drug product. A risk-based approach can (and should) be used to determine which elements need to be controlled. This approach can save your company significant amounts of time, money, and testing, but it requires some up-front effort in the form of a risk assessment.
Since the requirements have been introduced, we are seeing additional technologies brought forward as potential tools for testing elemental impurities, including laser ablation (which may be useful for samples with high silicon dioxide content), laser induced breakdown spectroscopy (LIBS), and X-ray fluorescence.
Risk Assessment Approach
Risk assessment for elemental impurities typically begins with obtaining information from your suppliers: Have they tested their raw materials for elemental impurities? Are these impurities controlled? Are they reported on the Certificate of Analysis (CoA)?
Specific groups of elements should be addressed, depending on the route of administration of the dosage form (Table 2). Generally, any element intentionally added and the four toxic elements (Cd, Pb, As, Hg) should be evaluated. Other elements are added based on potential toxicity associated with the route of administration.
Table 2: Elements to be Considered in Risk Assessment as a Function of Route of Administration

Adapted from International Conference on Harmonisation GUIDELINE FOR ELEMENTAL IMPURITIES Q3D Current Step 4 version (Dec. 16, 2014)
We recommend a systematic approach to risk assessment for elemental impurities:
- Identify which materials will be included in the drug product and the amount present in each dosage form (if multiple strengths, focus on that with the greatest risk, usually the highest potency).
- Collect information from the suppliers, including the drug substance and each excipient, including Certificates of Analysis and Supplier Control Statements, regarding elemental impurities.
- Create a table that displays the potential sources of elemental impurities, using information from the Certificates of Analysis. If a supplier does not provide a quantitative result, it may be necessary to assume the elemental impurities are present at the ICH limit. See Table 3 for an example. Review the table to determine the greatest risks.
- Verify the information on the suppliers’ Certificates of Analysis by actual testing of three distinct lots, which should be representative of what will be received in the future. This may be done in-house or at a contract testing laboratory that is qualified to test pharmaceutical materials for elemental impurities. (Note that some laboratories that test elemental impurities for food or environmental materials may not have appropriate sensitivity for this test or may not have suitable CGMPs in place.)
- If testing shows levels that are less than 30 percent of the permitted daily exposure (PDE), routine testing may not be necessary. Although the ICH Q3D Training Module 5 supports this approach, ultimately the decision is made by the regulatory agency reviewing the application.
- Based on the risk assessment, propose a testing plan that addresses which materials will be tested for which metals at what frequency.
Since the limits for the elements apply to the drug product, the user has some options. For instance, control may be demonstrated in the components (drug substance and excipients) or directly in the drug product. The USP describes both approaches. Many companies are adopting the component approach, since most elemental impurities enter the system via the components and since there is probably no way to recover if unacceptable levels of elemental impurities are observed in the finished drug product.
Sample Preparation
Sample preparation is an important consideration for testing elemental impurities. Since the metals must be in solution to be measured by ICP (the analytical technique recommended by USP in General Chapter <233>), the sample must be fully dissolved. For soluble materials, water or a solvent may be capable of dissolving the sample. Often, digestion in hot, strong acids is necessary to dissolve the sample. Acids may include nitric acid, hydrochloric acid, or (for some challenging samples) hydrofluoric acid. Acid digestion is often carried out in closed vessels, both to assure safety of the analyst and to reduce loss of volatile components. When using closed vessel digestion, always follow the manufacturer’s instructions to maintain a safe environment. Due to its volatility, mercury may pose particular problems for testing.
Risk Evaluation And Control Strategy
The outcomes from the risk assessment should be an evaluation of the risks and a control strategy, which will assure that future products will comply with the requirements.
Elemental impurity levels in components, shown in Table 3, indicate that among the four toxic metals (Class 1), based on review of supplier analysis and testing of representative lots, with the exception of the iron oxides, all of the levels are relatively low compared to the requirements. Even if we considered a daily dose of 10 g (used by the guidances as the likely maximum for most drugs), the amount of Pb and As would be below the ICH limit, and even below the control threshold of 30 percent of those limits (proposed by ICH).
Table 3: Evaluation of Elemental Impurities in Components (µg/g)
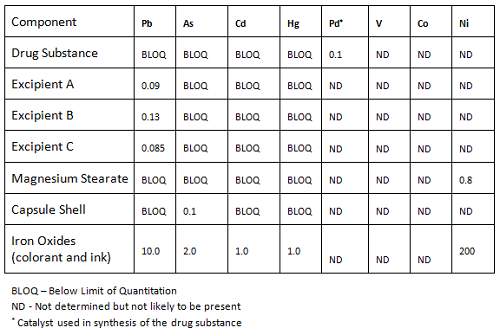
The levels of the Class 2A elements were not determined, since information from the supplier and review of the production for those materials concluded they were not likely to be present.
The iron oxides present a potential concern, due to the presence of all four of the Class 1 metals. This leads us to look at the amounts of this component used in the drug product.
Table 4 shows a summary of the amounts of the various components that will be used in the drug product and the associated levels of elements from Class 1 and 2A, as well as palladium, which is used in the synthesis of the drug substance. When the amounts of each element expected to be present in the drug product are summed, and those sums are compared to the control thresholds and ICH limits, it is clear the elemental impurities should not be a concern.
Table 4: Evaluation of Elemental Impurities in Drug Product
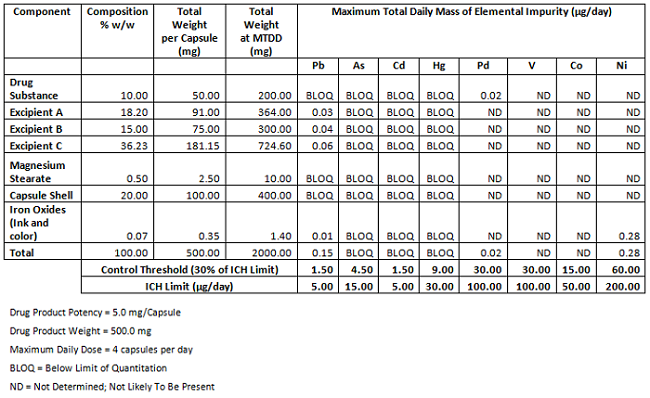
A logical conclusion from this evaluation for the control strategy would be that it is not necessary to control the elemental impurities in the components or drug product by routine testing. This is certainly a proposal that could be made in a regulatory filing. However, since the requirements have only recently been implemented, and regulatory agencies may not have extensive experience with enforcing these requirements, regulatory pushback is not unexpected. Firms may be asked to perform testing, at least for the Class 1 metals¸ until there is more experience. One approach a firm might take in this situation is to propose a “sunset specification”, in which the firm agrees to test a specified number of lots of either components or drug product, after which (if the preliminary information confirms levels are consistently below the control threshold) the requirement for testing could be reduced or eliminated.
Conclusion
The new elemental impurity requirements are now official and subject to regulatory review, including inspections. These requirements are much tighter than the historical heavy metal requirements and address a much broader array of elements. As a result, when testing is conducted, it will require more expensive testing procedures. Adopting a risk assessment approach to evaluate the impact and development of a science-based strategy for control of elemental impurities has the potential to significantly reduce the amount of testing that must be performed routinely, resulting in savings in testing, time, and costs.
When using a risk-based approach, it should be rigorous and well documented, including the rationale used to arrive at the testing scheme that will be used. At this point, since regulatory agencies are just getting familiar with the requirements and associated compliance, it can be expected that some negotiations will be necessary to come to mutually agreeable plan to demonstrate compliance.
About The Author:
 Greg Martin is president of Complectors Consulting, which provides consulting and training in pharmaceutical analytical chemistry. Martin has over 25 years of experience in the pharmaceutical industry and was director of pharmaceutical analytical chemistry (R&D) for Merck for a number of years. In addition, he has volunteered for the USP for over 10 years, and currently serves on the General Chapters – Chemical Analysis Expert Committee and the Expert Panels on Validation and Verification, Residual Solvents, and Use of Enzymes for Dissolution Testing of Gelatin Capsules. Martin is author of several papers in the areas of dissolution and analytical method validation, and is past chair of the AAPS In Vitro Release and Dissolution Testing Focus Group. He can be contacted at greg.martin@complectors.com.
Greg Martin is president of Complectors Consulting, which provides consulting and training in pharmaceutical analytical chemistry. Martin has over 25 years of experience in the pharmaceutical industry and was director of pharmaceutical analytical chemistry (R&D) for Merck for a number of years. In addition, he has volunteered for the USP for over 10 years, and currently serves on the General Chapters – Chemical Analysis Expert Committee and the Expert Panels on Validation and Verification, Residual Solvents, and Use of Enzymes for Dissolution Testing of Gelatin Capsules. Martin is author of several papers in the areas of dissolution and analytical method validation, and is past chair of the AAPS In Vitro Release and Dissolution Testing Focus Group. He can be contacted at greg.martin@complectors.com.